TRAIL
发布时间:2019-12-11 15:06 来源:SABiosciences
- 通路
- 概述
Review
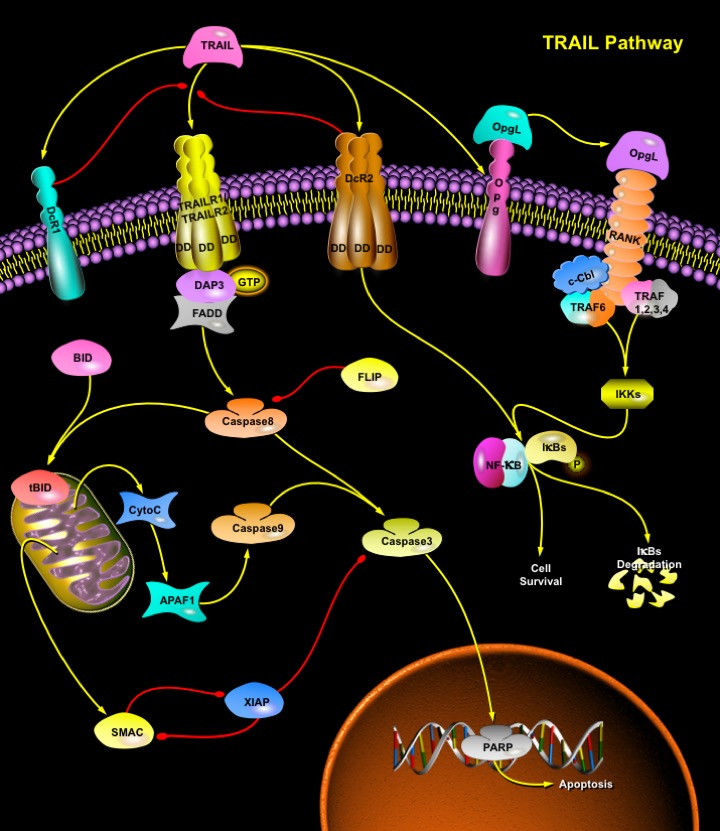
TRAIL (TNF-Related Apoptosis-Inducing Ligand) is a protein consisting of 281 amino acids. It is also called Apo2L. Five proteins, TRAILR1 (DR4), TRAILR2 (DR5/ TRICK2 or KILLER), TRAILR3 (DcR1/ TRID or LIT), TRAILR4 (DcR2 or TRUNDD), and Opg (Osteoprotegerin), have been identified as TRAIL receptors (Ref.1). Both TRAILR1 and TRAILR2 contain the functional DD (Death Domain), capable of inducing apoptosis. The other three receptors DcR1, DcR2 and Opg serve as "decoy" receptors. These three receptors can bind to TRAIL, but cannot induce apoptosis. DcR1 is a glycosylphosphatidylinositol-anchored cell surface protein, which contains the TRAIL-binding region as well as a region that anchors the receptor to the membrane. But unlike the other receptors, DcR1 lacks an intracellular tail needed to spark the death pathway. DcR2 is also able to bind TRAIL but contains a truncated DD that does not signal apoptosis induction but can activate NF-KappaB (Nuclear Factor-KappaB). Finally, Opg, slightly weaker receptor for TRAIL also binds to OpgL (Osteoprotegrin Ligand/Receptor), activator for NF-KappaB ligand (RANKL/Receptor Activator of NF-KappaB Ligand), inhibits osteoclastogenesis, and increases bone density. A cell expressing more decoy receptors is more likely to survive upon binding of the death ligands. TRAIL has been demonstrated to kill a wide variety of tumor cells with minimal effects on normal cells. This is because TRAIL's death receptors (TRAILR1 and TRAILR2) are mainly expressed in transformed cells while its decoy receptors are expressed in normal cells.
Binding of TRAIL to DR4 and DR5 leads to the recruitment of an adaptor protein FADD (Fas-Associated Death Domain), which functions as a molecular bridge to Caspase8, a protease at the apex of the cell death cascade (Ref.2). DR4 and DR5 have been proposed to indirectly bind FADD via the GTP-binding adaptor molecule, DAP3 (Death Associated Protein-3). Caspase8 then oligomerizes and is activated via autocatalysis. Activated Caspase8 stimulates apoptosis via two parallel cascades: it directly cleaves and activates Caspase3, and it cleaves BID (BH3 Interacting Death Domain) (Ref.3). tBID (Truncated BID) translocates to mitochondria, inducing CytoC (Cytochrome-C) release, which sequentially activates Caspases9 and 3. BAX (BCL-2 Associated X-Protein) deficiency has no effect on TRAIL-induced Caspase8 activation and subsequent cleavage of BID; however, it results in an incomplete Caspase3 processing due to inhibition by XIAP (Inhibitor of Apoptosis, X-Linked). Release of SMAC/DIABLO from mitochondria through TRAIL-Caspase8-tBID-BAX cascade is required to remove the inhibitory effect of XIAP and allow apoptosis to proceed. BAX-dependent release of SMAC, not CytoC from mitochondria, mediates the contribution of mitochondrial pathway to TRAIL through death receptor-mediated apoptosis. Caspase3 directly cleaves downstream substrates e.g. PARP (Poly ADP Ribose Polymerase).
Upon binding to TRAILR1, TRAILR2, or TRAILR4, TRAIL can also activate the transcriptional factor NF-KappaB and JNK (c-Jun N-terminal Kinase). Recently, it was suggested that TRAF2 (TNF Receptor-Associated Factor-2), an important effector of TNF (Tumor Necrosis Factor) signaling, was involved in both NF-KappaB and JNK activation induced by overexpression of TRAIL receptors. Overexpression of TRAF2 blocked TRAIL-induced IKKs (I-KappaB Kinases) and JNK activation; the absence of TRAF2 affected TRAIL-induced JNK activation but had little effect on IKK activation. In addition, neither RIP (Receptor-Interacting Protein) nor TRAF2 was required for TRAIL-induced apoptosis (Ref.5). In response to many stimuli such as TNF and IL-1 (Interleukin-1), the activation of NF-KappaB is mediated through IKK and JNK is activated through the MAPK (Mitogen-Activated Protein Kinase) cascade, namely, JNKK1 (MKK4) and MEKK1. The activation of JNK is a major regulatory step to activate the transcription factor Activatng Protein-1. Inactive NF-KappaB is located in the cytoplasm because its interaction with the inhibitory proteins, I-KappaBs, masks its nuclear translocation signal. When IKK is activated, it phosphorylates I-KappaBs. Then the phosphorylated I-KappaBs will be polyubiquitinated and rapidly degraded by the proteasome. The degradation of I-KappaBs leads to the release of NF-KappaB and allows NF-KappaB to translocate into the nucleus and to activate its target genes, some of which are the crucial mediators of the NF-KappaB antiapoptotic function. TRAIL is expressed on different cells of the immune system and plays a role in natural killer cell–mediated tumor surveillance. It exhibits specific tumoricidal activity against a variety of tumors. TRAIL can cause significant death of normal human hepatocytes. TRAIL is an important regulator of the IFN-dependent apoptotic process in melanoma and myeloma cell lines and in dendritic and natural killer cells.
References
- 1
- Petak I, Douglas L, Tillman DM, Vernes R, Houghton JA. Pediatric rhabdomyosarcoma cell lines are resistant to Fas-induced apoptosis and highly sensitive to TRAIL-induced apoptosis.
- 2
- Clarke P, Meintzer SM, Gibson S, Widmann C, Garrington TP, Johnson GL, Tyler KL. Reovirus-induced apoptosis is mediated by TRAIL.
- 3
- Gibson SB, Oyer R, Spalding AC, Anderson SM, Johnson GL. Increased expression of death receptors 4 and 5 synergizes the apoptosis response to combined treatment with etoposide and TRAIL.
- 4
- Lin Y, Devin A, Cook A, Keane MM, Kelliher M, Lipkowitz S, Liu ZG. The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of IkappaB kinase and c-Jun N-terminal kinase.
 关于我们
关于我们