CREB_Pathway
发布时间:2019-12-10 09:52 来源:SABiosciences
- 通路
- 概述
Review
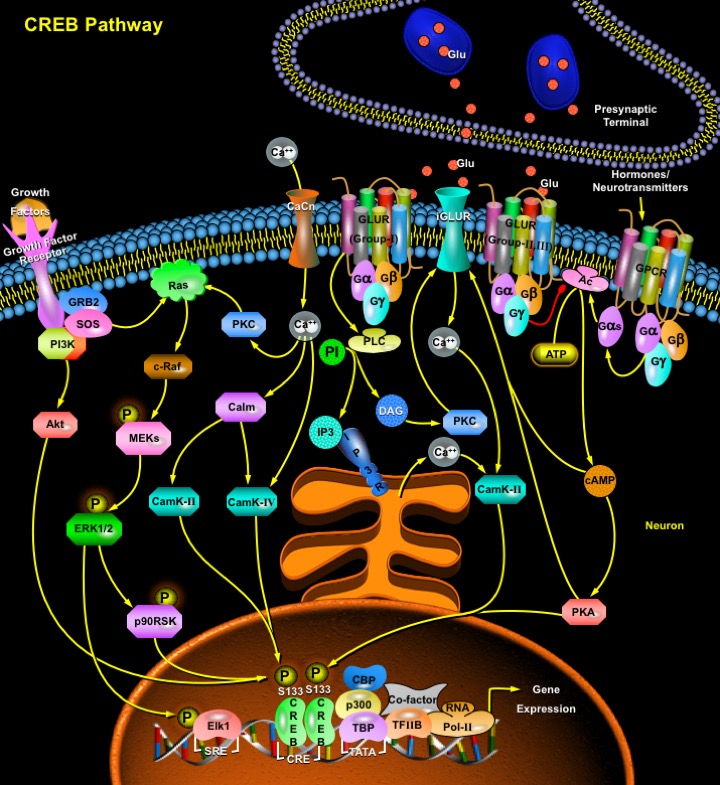
The process of consolidating a new memory and the dynamic complexity of information processing within neuronal networks is greatly increased by activity-dependent changes in gene expression within individual neurons. A leading paradigm of such regulation is the activation of the nuclear transcription factor CREB (cAMP Responsive Element Binding Protein), and its family members the ATF (Activating Transcription Factor) and CREM (cAMP Response Element Modulator), which belong to bZIP (basic/leucine zipper) class of transcription factors that functions in vivo to regulate the proliferation of pituitary cells and thymocytes. Proteins belonging to this class are characterized by the ability to bind to the consensus sequence TGACGTCA (Ref.1, 2 & 3) and contain a leucine zipper responsible for DNA binding (basic region) and for dimerization (leucine zipper region) of the proteins. CREB can form homodimers or heterodimers with other members of the ATF family, including ATF1 and CREM. However, heterodimerization of CREB with other members of the ATF family decreases its stability and CRE (cAMP Responsive Element) binding affinity (Ref.4).
Changing levels of cAMP, Ca2+ and TGF-Beta (Transforming Growth Factor-Beta) regulate CREB and its closely related proteins (SHC, GRB2, SOS, HRas, cRaf, etc) that implicate in a variety of biological responses such as neuronal excitation, long-term memory formation, neural cell proliferation, and opiate tolerance (Ref.5). Through interaction with its nuclear partner CBP (CREB Binding Protein), it drives the transcription of a large number of genes. Several different protein kinases possess the capability of driving this phosphorylation, making it a point of potential convergence for multiple intracellular signaling cascades. The crucial event in the activation of CREB is the phosphorylation of Ser133 in KID (Kinase-Inducible Domain). This domain includes several consensus phosphorylation sites for a variety of kinases like PKA (Protein Kinase-A), PKC (Protein Kinase-C), CSNK (Casein Kinases), CaMKs (Calmodulin Kinases), GSK3 (Glycogen Synthase Kinase-3) and p70S6K that can either increase or decrease the activity of CREB. Ser133 phosphorylation of CREB can be caused by electrical activity, Growth Factors, Neurotransmitter or Hormone action on GPCR (G-Protein-Coupled Receptors), or by Neurotrophin effects on RTKs (Receptor Tyrosine Kinases) (Ref.6). Upon stimulation of cellular GPCR (G-Protein-Coupled Receptors) and Growth Factor Receptors, AC (Adenylate Cyclase) is activated, by G-proteins: GN-Alpha, GN-Beta and GN-Gamma leading to increases in cAMP. This in turn activates PKA by dissociating the regulatory (PKAR) from the catalytic (PKAC) subunits. In the basal state, PKA resides in the cytoplasm as an inactive heterotetramer of paired regulatory and catalytic subunits. Induction of cAMP liberates the catalytic subunits. This activated PKAC then recruits the Ca2+/CalmK-IV (Calmodulin (Calm)-dependent Kinases), MEK (MAPK/ERK Kinases)/ ERK1/2 (Extracellular Signal-Regulated Kinases) and together they translocate to the nucleus (Ref.7 & 11). In the nucleus they lead to the recruitment of the transcriptional coactivators CBP (CREB Binding Protein) and p300 by phosphorylating Elk1. Elk1 is a part of a TCF (Ternary Complex Factor) that activates RSKs (Ribosomal S6 Kinases) and binds SRF (Serum Response Factor) to the SRE (Serum Response Element). Phosphorylation of Elk1 increases its transcriptional ability to form ternary complexes with SRF at the SRE in the promoter region of many genes, such as c-Fos (Ref.8). CBP/p300 stimulates gene expression by interacting with components of the general transcriptional machinery or by promoting the acetylation of specific lysine residues in nucleosomes located near transcriptionally active promoters thus creating access to the gene for the basal transcriptional machinery. The basal transcriptional machinery includes TBP (TATA-binding protein), TFIIB (Transcription Factor-II-B), and RNA Pol-II (RNA Polymerase-II) (Ref.9). The accumulation of cAMP in response to activation of GPCR also induces PLC-Gamma (Phospholipase-C-Gamma) that catalyzes the formation of DAG (Diacylglycerol), a PKC activator through PI (Phosphatidylinositols). PI3K (Phosphoinositide-3kinase) is responsible for activation of Akt/PKB (Protein Kinase-B) which directly or indirectly affects CREB.
In the presynaptic terminal, GLUR (metabotropic Glutamate Receptors Group-I) augment Glu (Glutamate) release via interaction of PKC and PKA whereas Group-II/III Receptors inhibit Glutamate release. Phosphorylation of Group-II/III Receptors (metabotropic Glutamate Receptors Group-II/III) also inhibits the transmitter release. These activities can indirectly regulate CREB and Elk1 phosphorylation in the postsynaptic neurons. In the postsynaptic striatal neurons, Group-I Receptors increase PKC activity as well as intracellular Ca2+ levels from internal store via PLC/DAG and PI/IP3 Pathways, respectively. Activated PKC induces an increase in extracellular Ca2+ influx through phosphorylation of iGluR (ionotropic Glutamate Receptors), in particular NMDARs (N-Methyl-D-Aspartate Receptors). Elevation of Ca2+ through CaCn (Calcium Channel) upregulates Ca2+-dependent CaMK-II/ ERK1/2 signaling cascades resulting in CREB and Elk1 phosphorylation. In contrast, Group-II/III Receptors suppress the Ca2+ cascades by inhibiting AC coupling to GPCRs such as Dopamine Receptors. The decreased cAMP level reduces PKA-dependent phosphorylation of NMDARs (Ref.10).
CREB can be phosphorylated at a number of sites other than Ser133, including Ser129, Ser142 and Ser143. Phosphorylation of Ser142 and dephosphorylation of Ser133 residue by CalmK represses CREB activity. Calcineurin dependent PP1 and PP2A (Protein Phosphatases) is involved in the dephosphorylation of CREB. In addition to dephosphorylation, repressors can also block CREB activity (Ref.9). The activation of plasma-membrane channels, including NMDARs and L-VGCCs also relieve repressor factors such as DREAM (Downstream Response Element (DRE)-Antagonist Modulator), and induce other activators like SRF, that work with CREB to drive c-Fos transcription. By contrast, only stimuli that elevate intracellular Ca2+ (by NMDAR and L-VGCC activation) lead to the phosphorylation of CREB at Ser142 and Ser143, and activate the transcription factor CaRF (Calcium Response Factor). Phosphorylation of Ser142/143 in CREB inhibits the association of phosphorylated Ser133 with CBP. CaRF cooperates with CREB to promote transcription of BDNF (Brain-Derived Neurotrophic Factor), assisting in cofactor recruitment and mediation of stimulus-selective gene transcription (Ref.8).
The cAMP/CREB signaling pathway has been strongly implicated in the regulation of a wide range of biological functions such as growth factor-dependent cell proliferation and survival, glucose homeostasis, spermatogenesis, circadian rhythms and the synaptic plasticity that is associated with a variety of complex forms of memory including spatial and social learning indicating that CREB may be a universal modulator of processes required for memory formation (Ref.6). Deletion of CREB and CREM in neurons of the developing CNS (Central Nervous System) results in apoptosis, and postnatal ablation of these genes results in neuronal degeneration in adulthood. Neurons of the adult striatum and hippocampus are particularly vulnerable to CREB/CREM deficiency. The richness of CREB signaling is greatly increased by its responsiveness to multiple intracellular signal transduction cascades and the potential for this family of transcription factors to induce and suppress gene expression renders them ideally suited for regulating gene expression during the process of epidermal differentiation (Ref.4).
References
- 1
- Josselyn SA, Nguyen PV. CREB, synapses and memory disorders: past progress and future challenges.
- 2
- Carlezon WA Jr, Duman RS, Nestler EJ. The many faces of CREB.
- 3
- . Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway.
- 4
- Johannessen M, Delghandi MP, Seternes OM, Johansen B, Moens U. Synergistic activation of CREB-mediated transcription by forskolin and phorbol ester requires PKC and depends on the glutamine-rich Q2 transactivation domain.
- 5
- Wu GY, Deisseroth K, Tsien RW. Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway.
- 6
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB.
- 7
- Choe ES, Wang JQ. Regulation of transcription factor phosphorylation by metabotropic glutamate receptor-associated signaling pathways in rat striatal neurons.
- 8
- Finkbeiner S. New roles for introns: sites of combinatorial regulation of Ca2+- and cyclic AMP-dependent gene transcription.
- 9
- Chawla S, Bading H. CREB/CBP and SRE-interacting transcriptional regulators are fast on-off switches: duration of calcium transients specifies the magnitude of transcriptional responses.
- 10
- Boulware MI, Weick JP, Becklund BR, Kuo SP, Groth RD, Mermelstein PG. Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein.
 关于我们
关于我们