VEGF_Pathway
发布时间:2019-12-11 15:29 来源:SABiosciences
- 通路
- 概述
Review
The formation of blood vessels occurs either by in situ differentiation of endothelial cell precursors (Angioblasts) and association of these cells to form primitive vessels, a process called Vasculogenesis, or by growth of preexisting vessels, a process called Angiogenesis. Vasculogenesis establishes the primary vascular plexus of the early embryo, whereas development of blood vessels during later embryogenesis and adult life occurs primarily by Angiogenesis. Angiogenesis is an integral feature of capillary sprouts from preexisting blood vessels. It is typically quiescent in the adult, except for pathological situations (e.g., wound healing, Diabetic Retinopathy, Rheumatoid Arthritis, Cardiac Ischemia, Psoriasis, tumor growth) and during menstrual cycle-specific physiological processes in the female reproductive system (e.g., ovulation, endometrial growth, implantation, placentation) (Ref.1 & 2). Once blood vessels have been established, endothelial cells undergo tissue-specific changes to generate numerous types of functionally distinct vessels as organs differentiate. These processes require that endothelial cells respond to a variety of extracellular signals that activate receptors responsible for growth and differentiation (Ref.3). VEGF (Vascular Endothelial Growth Factor), Angiopoietin and Ephrin are key molecules in the promotion of angiogenesis via activation of the VEGFR (VEGF Receptor), TIE and Ephrin expressed on vascular endothelial cells, respectively (Ref.4). VEGF signaling plays a major role in promoting the proliferation and differentiation of the endothelial lineage from the earliest stages of development, whereas the Angiopoietin/TIE2 pathway acts slightly later to promote the recruitment of supporting cells and vessel stabilization. However, the complex patterning of the developing vasculature also requires specific vascular roles for the components of other signaling pathways, including the Ephrin, TGF-Beta (Transforming Growth Factor- Beta), PDGF (Platelet-Derived Growth Factor), FGF (Fibroblast Growth Factor), and Delta-Notch Pathways (Ref.6).
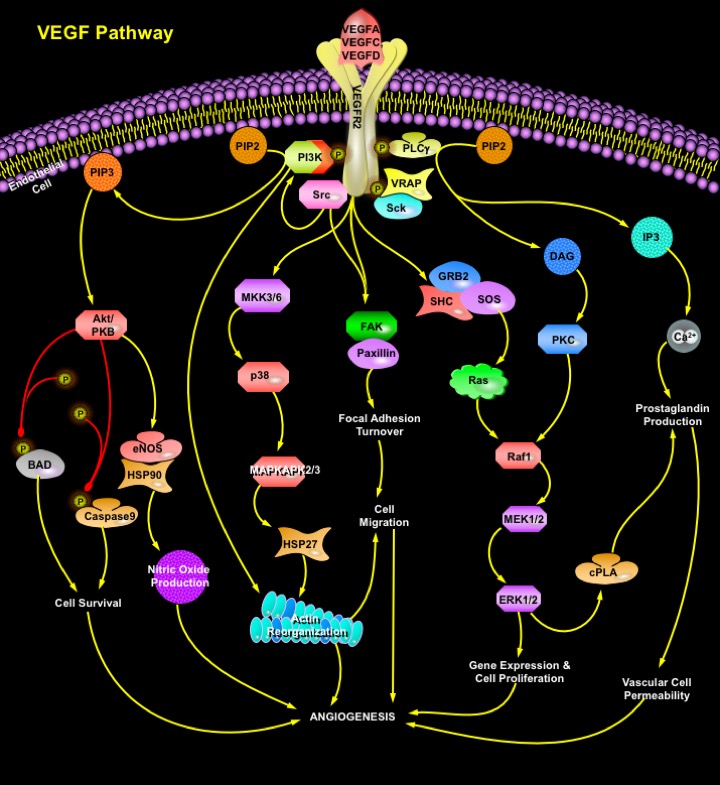
VEGF is a heparin-binding homodimeric glycoprotein that acts via endothelial-specific receptor tyrosine kinases, VEGFR1 (Flt1), VEGFR2 (KDR/Flk1), and VEGFR3 (Flt4) (Ref.3). Besides VEGFA, the VEGF family of growth factors currently contains five other known members, namely PlGF (Placenta Growth Factor), VEGFB, VEGFC, VEGF-D and orf viral VEGF homologs. Additional novel VEGF-like heparin-binding proteins have been isolated recently from snake venom. Disruption of the genes encoding either VEGF or any of the three receptors of the VEGF family, results in embryonic lethality because of failure of blood vessel development. VEGFR2 is the main signal transducing VEGF receptor for angiogenesis and mitogenesis of endothelial cells. After receptor dimerization and autophosphorylation, several SH2 domain-containing signal transduction molecules are activated either directly such as PLC-gamma, VRAP (VEGF Receptor-Associated Protein), and Sck, or by indirect mechanisms, such as Src and PI3K (Phosphatidylinositol 3-Kinase). Activation of PKC (Protein Kinase-C) plays a crucial role in VEGFA mitogenic signaling via the Raf1-MEK-ERK pathway. Cell survival signal is mainly mediated through PI3K-mediated activation of Akt/PKB (Protein Kinase-B). Activation of PI3K results in accumulation of PIP3 (Phosphatidylinositol-3, 4, 5-Trisphosphate), which in turn mediates membrane targeting and phosphorylation of Akt/PKB by binding to its PH (Pleckstrin Homology) domain. Downstream targets for Akt/PKB pathway include the proapototic proteins BAD, FKHR1 (Forkhead Transcription Factor-1), and Caspase-9, whose phosphorylation inhibits apoptosis. Moreover, VEGFA induces expression of the antiapoptotic protein BCL2 and the IAP (Inhibitor of Apoptosis Protein) family members XIAP (Xenopus Inhibitor of Apoptosis), and Survivin in HUVEC (Human Umbilical Vein Endothelial) cells, suggesting that these proteins also play an important role in endothelial cell survival (Ref.5). PLC-Gamma catalyzes the hydrolysis of PIP2 (Phosphatidylinositol-4, 5-Bisphosphate), creating IP3 (Inositol Trisphosphate) and DAG (Diacylglycerol), which stimulate the release of Ca2+ from internal stores and activate PKC. VEGF-A-induced Ca2+ mobilization is involved in short-term production of Nitric Oxide and Ptg (Prostaglandin). SHC phosphorylation promotes formation of SHC-GRB2 (Growth Factor Receptor-Bound Protein-2)-SOS complexes and induces PKC-dependent and Ras-independent induction of the Raf1-MEK-ERK1/2 pathway in sinusoidal endothelial cells and in HUVEC cells (Ref.5). Negative feedback for the mitogenic effects of VEGF is provided by cPLA2 (cytosolic Phospholipase-A2), activation and biosynthesis of Prostaglandin. p38 pathway conveys the VEGF signal to microfilaments inducing rearrangements of the Actin cytoskeleton that regulate endothelial cell migration by modulating the activation of MAPKAPK2/3 (MAP Kinase Activated Protein Kinase-2/3) and phosphorylation of the F-Actin polymerization modulator, HSP27 (Heat Shock Protein-27). The activation of FAK (Focal Adhesion Kinase) and Paxillin by VEGFA in HUVE cells through VEGFR2 leads to recruitment of Actin-anchoring proteins such as Talin and Vinculin to the focal adhesion plaque, which are essential for VEGFA-induced actin reorganization. VEGFA also stimulates tyrosine phosphorylation of the FAK-related cytoplasmic tyrosine kinase PYK2 (also termed RAFTK, "Related Adhesion Focal Tyrosine Kinase") in a bone marrow endothelial cell line (Ref.5).
Although VEGF is known to be a powerful growth factor for therapeutic angiogenesis/vascularization in the ischemic hind limb and myocardium, it has other activities that can increase the proliferation and permeability of capillary endothelial cells. These activities may produce unwanted side effects, such as tumor angiogenesis, vascular leakage, edema, and inflammation (Ref.9). Different cytokines including VEGFA, have been reported to modulate Kaposi's Sarcoma (KS), a major vascular tumor commonly associated with HIV1 (Human Immunodeficiency Virus) and HHV-8 (Human Herpes Virus) (Ref.7 & 8). In endothelial cells, the VEGF-Flk1/KDR signal system is a very important generator of NO (Nitric Oxide) through the activation of its downstream effectors PI3K, Akt kinase and eNOS (endothelial NO Synthase). NO regulates hematopoiesis and modulates AML (Acute Myeloid Leukaemia) cell growth (Ref.10). VEGF also play a major role in the progression of ovarian cancer and colon cancer by modulating tumor proliferation through its promotion of tumor angiogenesis. In a variety of human pathological situations that are associated with aberrant endothelial proliferation and aberrant neovascularization VEGF-based therapy to treat diseases include the use of neutralizing antibodies against VEGFA or VEGFR2, antisense oligonucleotides, negative regulatory peptides, soluble receptors, and ATP analogs to inhibit the kinase activity of VEGFR (Ref.5). However, inhibition of VEGF function may result in infertility by blockade of Corpus Luteum function. Interference with VEGF function has therefore become of major interest for drug development to block angiogenesis and targeting the VEGF signaling pathway may be of therapeutic importance for many diseases.
References
- 1
- Mueller MD, Vigne JL, Minchenko A, Lebovic DI, Leitman DC, Taylor RN. Regulation of vascular endothelial growth factor (VEGF) gene transcription by estrogen receptors alpha and beta.
- 2
- Voelkel NF, Vandivier RW, Tuder RM. Vascular endothelial growth factor in the lung.
- 3
- Shida A, Fujioka S, Ishibashi Y, Kobayashi K, Nimura H, Mitsumori N, Suzuki Y, Kawakami M, Urashima M, Yanaga K. Prognostic significance of vascular endothelial growth factor D in gastric carcinoma.
- 4
- Endo A, Nagashima K, Kurose H, Mochizuki S, Matsuda M, Mochizuki N. Sphingosine 1-phosphate induces membrane ruffling and increases motility of human umbilical vein endothelial cells via vascular endothelial growth factor receptor and CrkII.
- 5
- Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction.
- 6
- Uyttendaele H, Ho J, Rossant J, Kitajewski J. Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium.
- 7
- Ganju RK, Munshi N, Nair BC, Liu ZY, Gill P, Groopman JE. Human immunodeficiency virus tat modulates the Flk-1/KDR receptor, mitogen-activated protein kinases, and components of focal adhesion in Kaposi
- 8
- Rusnati M, Urbinati C, Musulin B, Ribatti D, Albini A, Noonan D, Marchisone C, Waltenberger J, Presta M. Activation of endothelial cell mitogen activated protein kinase ERK(1/2) by extracellular HIV-1 Tat protein.
- 9
- Chae JK, Kim I, Lim ST, Chung MJ, Kim WH, Kim HG, Ko JK, Koh GY. Coadministration of angiopoietin-1 and vascular endothelial growth factor enhances collateral vascularization.
- 10
- Koistinen P, Siitonen T, Mantymaa P, Saily M, Kinnula V, Savolainen ER, Soini Y. Regulation of the acute myeloid leukemia cell line OCI/AML-2 by endothelial nitric oxide synthase under the control of a vascular endothelial growth factor signaling sys
 关于我们
关于我们