Transendothelial_Migration_of_Leukocytes
发布时间:2019-12-11 15:19 来源:SABiosciences
- 通路
- 概述
Review
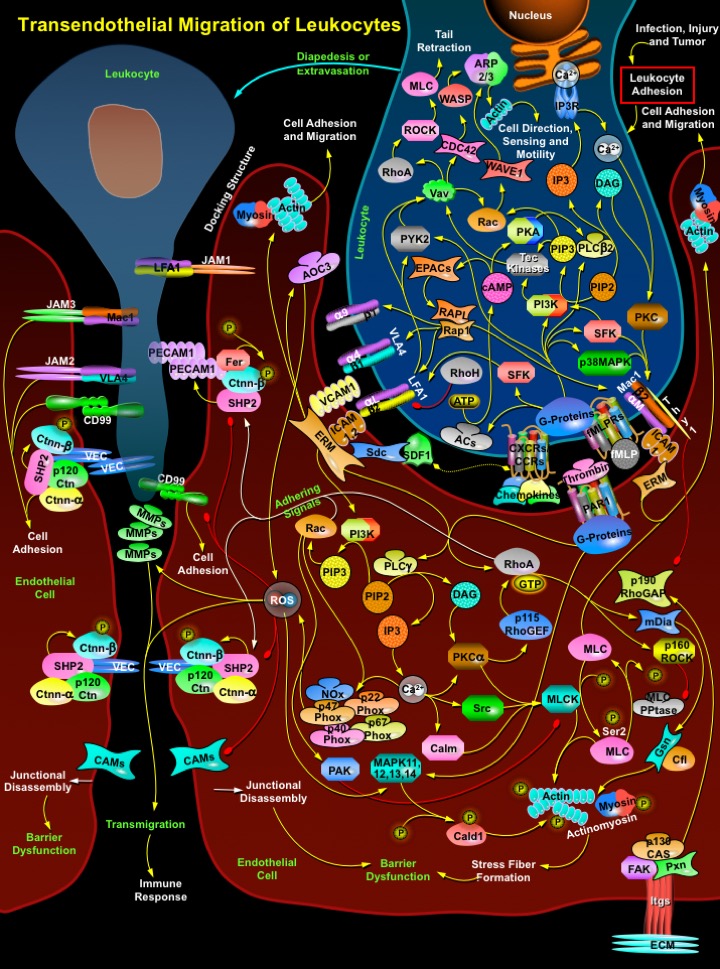
The migratory properties of leukocytes or WBCs (White Blood Cells) are indispensable to drive immune responses throughout the body. Leukocytes fall under two categories according to their morphology that includes Agranulocytes (Lymphocytes and Monocytes) and Granulocytes (Neutrophils, Basophils and Eosinophils). To ensure migration to the proper locations, the trafficking of leukocytes is tightly regulated. The vascular endothelium lining the intima of the blood vessels (both in lymphoid and non-lymphoid tissues) regulates a variety of functions including vascular smooth muscle tone, host-defense reactions, Angiogenesis, and tissue fluid hemostasis. The maintenance by the endothelium of a semi-permeable barrier is particularly important in controlling the passage of macromolecules and fluid between the blood and interstitial space. It is known that loss of this function results in tissue inflammation, the hallmark of inflammatory diseases such as the acute respiratory distress syndrome. The characteristic permeability of transported macromolecules is dependent on their molecular radii as well as the barrier properties of the particular endothelium (Ref.1 & 2). The Extravasation of leukocytes is essential for many (patho) physiological processes, including migration of T-lymphocytes for immune surveillance, recruitment of activated lymphocytes and granulocytes during acute and chronic inflammatory responses, and homing and mobilization of hematopoietic progenitor cells. The Transmigration and Extravasation of leukocytes across the endothelium that lines the vessel wall occurs in several distinct steps, referred to as the multi-step paradigm. The first step comprises the Rolling of the leukocytes over the endothelial cells, mediated by transient weak interactions between adhesion molecules. Subsequently, loosely attached leukocytes are in such close proximity of the endothelium that they are activated by chemotactic cytokines, presented on the apical surface of the endothelium. As a consequence, the activated leukocytes will spread and firmly adhere to the endothelium forming docking structures and finally migrate through the intercellular clefts between the endothelial cells to the underlying tissue (Ref.3 & 4).
In leukocytes, Chemokines and fMLP (N-formyl-Met-Leu-Phe) and their receptors CXCRs (Chemokine (C-X-C) Receptors)/CCRs (Chemokine-CC-Motif Receptors)) and fMLPRs (fMLP Related Receptors), respectively; are one of the many levels that coordinate the migration at various levels, resulting in a tightly controlled and very complex system of cell trafficking. Chemokines transmit their pro-migratory signals through these GPCRs (G-Protein-coupled Membrane Receptors). These receptors initiate adhesion and motility via G-proteins, leading to Integrin activation via inside-out signaling, followed by coordinated Actin polymerization, spreading at the leading edge of the cell and contraction at the back (Ref.5). Concomitant activation of the CXCRs/CCRs and fMLPRs at the cell surface is relayed to the G-proteins that in turn activate Ras, transmembrane AC (Adenylyl Cyclase), PLC-Beta2 (Phospholipase-C-Beta2); and protein kinases like SFKs (Src Family Tyrosine Kinases), PI3K (Phosphatidylinositde-3 Kinase), p38MAPK (p38 Mitogen-Activated Protein Kinase) and PYK2 (Proline-Rich Tyrosine Kinase-2) that leads to the polarization of the cell body and of the Actin cytoskeleton. The transmembrane ACs generates cAMP (Cyclic Adenosine 3',5'-monophosphate) from ATP (Adenosine Triphosphate). Here cAMP targets PKA (cAMP dependent Protein Kinase-A) and the GTP-exchange protein, EPACs (Exchange Protein Activated by cAMP). EPACs activate Rap1 (Ras-Related Protein-1) and RAPL (Regulator for Cell Adhesion and Polarization Enriched in Lymphoid Tissue) that forms tertiary complexes with Integrins like LFA1 (Leukocyte Function-Associated Antigen-1), a complex of Itg-AlphaL (Integrin-Alpha-L) and Itg-Beta2 (Integrin-Beta-2); Itg-Alpha9/Itg-Beta1; VLA4 (Very Late Antigen-4), comprising of Itg-Alpha4 and Itg-Beta1; and Mac1, which includes Itg-AlphaM and Itg-Beta2. cAMP stimulated PKA activates PLC-Beta2, which cleaves PIP2 (Phosphatidylinositol 4,5-bisphosphate) to generate DAG (Diacylglycerol) and IP3 (Inositol 1,4,5-trisphosphate). IP3 modulates release of Ca2+ (Calcium) through IP3R (IP3 Receptor) and along with DAG it facilitates activation of PKC (Protein Kinase-C). The PKC, SFKs and p38MAPK in turn activate Mac1 and LFA1 (Ref.6 & 7).
These activated Integrins bind to members of the Immunoglobulin superfamily that includes the important ones like ICAM1 (Intercellular Adhesion Molecule-1), ICAM2, VCAM1 (Vascular Cell Adhesion Molecule-1) and Thy1 (Thy1 Cell Surface Antigen) on the endothelial cells. This causes tight adherence of leukocytes to the endothelium. Adhesion via activated Integrins is a prerequisite for the active migration of leukocytes on the endothelial cell surface and finally through the layer of endothelial cells. Clustering of Integrins with Immunoglobulin superfamily members induces RhoA activation and corresponding stress fiber formation. Thus events in the leukocyte are regulated by two major classes of signaling molecules that are Integrins, which bind to stationary ligands on endothelial cells, and the GPCRs, which are activated by chemokines. This mechanism enables quick responses like the reorganization of the cytoskeleton, which is regulated by Rho GTPases. Rho GTPase contribution to leukocyte-endothelial interactions is mainly focused on the three founding members: RhoA, Rac (Ras-Related C3 Botulinum Toxin Substrate), and CDC42 (Cell Division Cycle-42). But RhoH activity maintains LFA1 in an inactive state until an appropriate signal triggers an adhesion-activating pathway such as that involving Rap1 (Ref.8 & 9). RhoH is a potent inhibitor of LFA1 adhesion to ICAM1. Under G-protein signaling PI3K enhances Tec Kinases and PIP3 (Phosphatidylinositol-3,4,5-Trisphosphate) activity in order to facilitate activation of Rac and Vav proteins. The activation of PYK2 (Proline-Rich Tyrosine Kinase-2), which is the result of a combination of signaling from both chemokine molecules and the Integrin ligands, enables it to form complexes with Vav proteins. Vav phosphorylation in turn triggers activation of RhoGTPases. Activated CDC42, Rac and RhoA bind to and specifically activate their downstream effectors, which are kinases such as ROCK (Rho-Associated Coiled-Coil-Containing Protein Kinase), scaffolding proteins like, WASP (Wiskott-Aldrich Syndrome Protein) and WAVE1 (WASP-family Verprolin-Homologous Protein). This allows signals flowing through Rho GTPases to coordinate the initiation of new Actin filaments through WASP and Actin-related protein, ARP2/3 complex and capacitate the leukocytes to sense cell direction. Again ROCK activation leads to an increase of MLC (Myosin Light Chain) activity leading to tail retraction and increased mobility (Ref.8).
On the endothelial side, often the chemokines like the SDF1 (Stromal Cell-Derived Factor-1) are presented and immobilized by Sdcs (Syndecans), the cell surface proteoglycans of the endothelium. SDF1 not only helps in the polarization of the leukocyte cell body but also enhances Integrin binding to Immunoglobulin superfamily members and transmit signals inside the endothelial cell via ERM (Ezrin, Radixin, and Moesin) proteins. ERM proteins are responsible for the formation of Actin and Myosin rich docking structures. These structures also possess clustered VCAM1, ICAM1, or both, Thy1 and the ERM proteins. Although this docking structure possibly facilitates leukocyte transendothelial migration, it is not necessary for firm adhesion of leukocytes to the endothelium. Hence further adhesion between PECAM1 (Platelet Endothelial Cell Adhesion Molecule-1), CD99 (CD99 Antigen) and attachment of LFA1, VLA4, Mac1 to JAM1 (Junctional Adhesion Molecule-1), JAM2, JAM3, respectively, are essential for firm adhesions (Ref.10 & 11). Cross-linking of Integrins and agonists such as Thrombin (through PAR1 (Protease Activated Receptor-1) activated G-proteins) induce increased intracellular Ca2+ levels by activation of PLC-Gamma leading to cleavage of PIP2 and formation of DAG and IP3. Increased intracellular Ca2+ also activates protein kinases such as PKC-Alpha (that requires DAG) or Src (v-Src Avian Sacroma (Schmidt-Ruppin A-2) Viral Oncogene) and Calm (Calmodulin). PKC-Alpha phosphorylates p115RhoGEF, which in turn increase RhoA GTPase activity. RhoA activates p160-ROCK (p160-Rho-Associated Coiled-Coil-Containing Protein Kinase) that regulates the state of Actin polymerization. p160-ROCK inhibits MLC PPtase (MLC Phosphatase) by phosphorylation and therefore increase levels of MLC-P (phosphorylated MLC at Ser2 (Serine-2)). Together, these events lead to increased Actin-Myosin cross-bridging (Actinomyosin formation), resulting in endothelial contraction or barrier dysfunction due to disassembly of junction proteins. RhoA also actives mDia that regulates Actin binding proteins like Gsn (Gelsolin) and Cfn (Cofilin) and alters Actin polymerization during Actinomyosin complex formation (Ref.10 & 12).
Contraction of endothelial cells thus leads to disassembly of adherens junctions, depolymerization of microtubules, and FAK (Focal Adhesion Kinase) activation. The stress fiber induced activation of FAK increases focal adhesion formation and promotes the interaction of endothelial cells with Integrins, ECM (Extracellular Matrix) proteins and other cytoskeletal modulators like p130CAS (Crk-Associated Substrate-P130) and Pxn (Paxillin). FAK also suppress RhoA activity by activating p190RhoGAP (p190-Rho GTPase Activating Protein) and triggers assembly of cell junction proteins. However, Thy1 expressed on the apical side of endothelial cells activate RhoA by decreasing the activity of p190RhoGAP to facilitate leukocyte transmigration (Ref.13). Under normal circumstances MLC promotes cell adhesion but upon stimulation by Src and Calm the activity of endothelial MLCK (Myosin Light Chain Kinase) increase and so the phosphorylation levels of MLC (MLC-P) and, subsequently, leading to the stimulation of Actinomyosin ATPase activity. However Rac induced PAK (p21/CDC42/Rac1-Activated Kinase) activation inhibits MLCK. Thrombin induced stress fiber formation also occurs through direct modulation of RhoA or by activation of MAPKs (Mitogen-Activated Protein Kinases) that phosphorylate Cald1 (Caldesmon-1). Cald1 increases stress fiber formation and induces contraction in the absence of MLC phosphorylation (Ref.8). Similarly, RhoA and Rac relay signals to Shp2 (Tyrosine Phosphatase Shp2) and p120Ctn/Ctnn-Delta1 (Catenin (Cadherin-Associated Protein)-Delta-1) to promote cell adhesion. It is known that PECAM1 function as a reservoir for tyrosine-phosphorylated Ctnn-Beta (Catenin-Beta) and PECAM1 binding to Ctnn-Beta prevents cytosolic Ctnn-Beta from degradation by the Proteosomes. But PECAM1 engagement induces Shp2 phosphorylation through Fer (Fps/Fes-Related Tyrosine Kinase), which then dephosphorylates PECAM1-bound Ctnn-Beta (Ref.1). Increased tyrosine phosphorylation of junctional proteins leads to loss of cell-to-cell adhesion and that sub-confluent endothelial monolayers have increased levels of tyrosine phosphorylation of the junctional proteins VEC (Vascular Endothelial Cadherin), Ctnn-Beta, Ctnn-Alpha (Catenin (Cadherin-Associated Protein)-Alpha) and p120Ctn. Moreover, Shp2 and Ctnn-Beta are both present in the VEC complex at cell-to-cell junctions. The Shp2 phosphatase activity is inhibited by ROS (reactive Oxygen Species), especially by Hydrogen Peroxide, because ROS mediates the oxidation of critical Cysteine residues in tyrosine phosphatases. As a result, tyrosine phosphorylation levels are increased, triggering loss of endothelial cell-to-cell junctions (Ref.14).
ROS are spontaneously generated by ERM protein interactions with AOC3 (Amine Oxidase Copper Containing-3)/VAP1 (Vascular Adhesion Protein-1). AOC3 catalyzes oxidative deamination and produces Aldehyde groups, Hydrogen Peroxide, and Ammonium. They are also generated when Ca2+ levels and Rac proteins upregulate Nox-based NADPH-oxidases like NOx1 (NADPH Oxidase-1), NOx2, NOx3; cytosolic factors like p47Phox, p67Phox, p40Phox; and CyB-Alpha (Cytochrome-B-245-Alpha Polypeptide)/p22Phox. ROS inhibits functioning of junction proteins like Shp2 and CD99 (CD99 Antigen) and also regulates activation of stress induced MAPKs and MMPs (Matrix Metalloproteinases) to promote stress fiber formation and degradation of VEC and other CAMs (Cellular Adhesion Molecules), respectively. This leads to the opening of inter-endothelial cell contacts, thus allowing leukocytes to transmigrate between adjacent endothelial cells to reach the underlying tissue (Ref.15 & 16). Interactions between activated leukocytes and endothelial cells demonstrate the interplay between cell-surface molecules, intracellular signaling cascades, and the control of motility and cell-cell adhesion. Understanding the complex series of events involved in cell-cell interactions during leukocyte transendothelial migration is a prerequisite for designing novel therapies to treat clinical conditions in which an inappropriate inflammatory response leads to disease. For such reasons leukocyte transmigration and adhesion represent important areas for drug targeting (Ref.17).
References
- 1
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability.
- 2
- van Buul JD, Hordijk PL. Signaling in leukocyte transendothelial migration.
- 3
- Nourshargh S, Marelli-Berg FM. Transmigration through venular walls: a key regulator of leukocyte phenotype and function.
- 4
- Engelhardt B, Wolburg H. Transendothelial migration of leukocytes: through the front door or around the side of the house?
- 5
- Rickert P, Weiner OD, Wang F, Bourne HR, Servant G. Leukocytes navigate by compass: roles of PI3Kgamma and its lipid products.
- 6
- Heit B, Colarusso P, Kubes P. Fundamentally different roles for LFA-1, Mac-1 and {alpha}4-integrin in neutrophil chemotaxis.
- 7
- Wittchen ES, van Buul JD, Burridge K, Worthylake RA. Trading spaces: Rap, Rac, and Rho as architects of transendothelial migration.
- 8
- Rolfe BE, Worth NF, World CJ, Campbell JH, Campbell GR. Rho and vascular disease.
- 9
- Hordijk P. Endothelial signaling in leukocyte transmigration.
- 10
- Yang L, Kowalski JR, Zhan X, Thomas SM, Luscinskas FW. Endothelial Cell Cortactin Phosphorylation by Src Contributes to Polymorphonuclear Leukocyte Transmigration In Vitro.
 关于我们
关于我们