TGF_Beta_Pathway
发布时间:2019-12-11 14:51 来源:SABiosciences
- 通路
- 概述
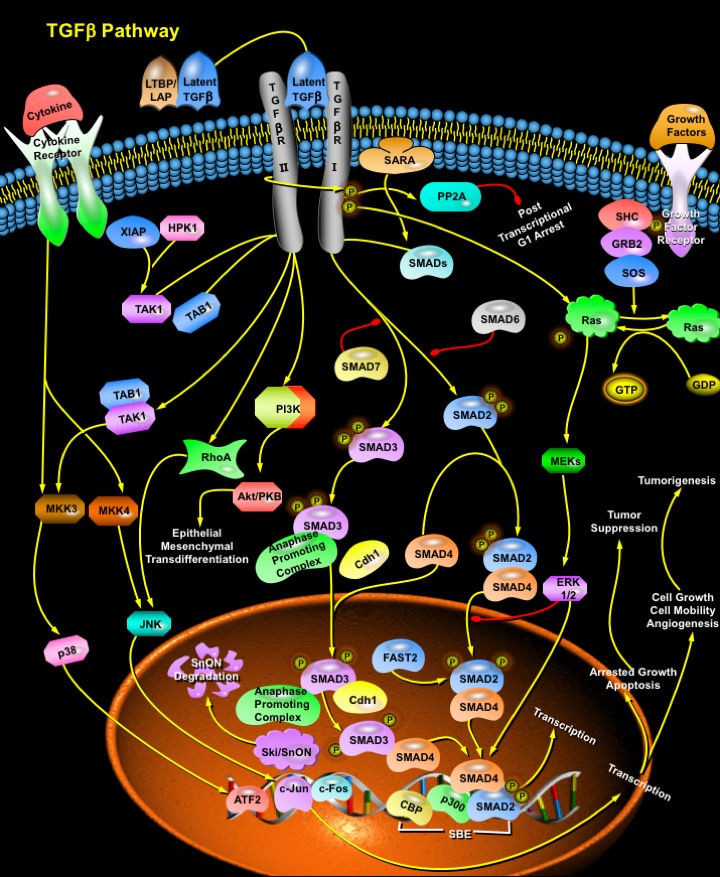
Review
Cell proliferation in somatic tissues, specification of cell fate during embryogenesis, differentiation and cell death are controlled by a multitude of cell–cell signals and loss of this control has devastating consequences. Prominent among these regulatory signals is the TGF-Beta (Transforming Growth Factor) super family, which comprises a large and diverse group of polypeptide morphogens including the prototype of the family–the TGF-Beta themselves as well as the BMPs (Bone Morphogenetic Proteins), and the GDFs (Growth and Differentiation Factors) (Ref.1). The members of the TGF-Beta family are expressed in distinct temporal and tissue-specific patterns and therefore play an important role in the development, homeostasis and repair of most tissues in organisms. All immune cell lineages, including B-Cell, T-Cell and dendritic cells as well as macrophages secrete TGF-Beta, which negatively regulates their proliferation, differentiation and activation by other cytokines. Thus, TGF-Beta is a potent immunosuppressor and perturbation of TGF-Beta signaling is linked to autoimmunity, inflammation and cancer (Ref.2).
Before binding to its receptors, TGF-Beta is activated from a large latent complex comprised of LTBP and LAP (Latency Associated Peptide). Ligand binding to the Type II receptor (TGF-BetaRII) leads to recruitment of Type I receptor (TGF-BetaRI) in the highly conserved juxtamembrane region, known as the GS domain. The activated TGF-BetaRI then phosphorylates its downstream targets, the members of the SMAD (Sma and Mad Related Family) family of signal transducers, SMAD2 and SMAD3 (Ref.3). They form hetero-oligomeric complexes with SMAD4 and translocate to the nucleus, where they interact at the promoter with other transcription factors at DNA sequence-specific binding sites ATF2 (Activating Transcription Factor-2) and SBE (SMAD Binding Element) to regulate gene expression. The heteromeric SMAD complex also interacts with transcriptional coactivators and corepressors like p300 and CBP (CREB Binding Protein) to connect the SMAD-TF complex with the basal transcriptional machinery and mediate the biological effects of TGF-Beta (Ref.3). SMAD2-SMAD4 complexes regulate transcriptional responses by specifically interacting with DNA-binding proteins such as FAST2 (Forkhead Activin Signal Transducer-2). Some of the activated target genes stimulate tumorigenesis while others suppress tumorigenesis. SMAD2 is multiply ubiquitinated later and selectively targeted for proteasome-dependent degradation (Ref.5). Over expression of SMAD7 inhibits phosphorylation of SMAD2 and SMAD3 by activated TGF-BetaRI. SMAD6 is quite different in structure from the other SMAD proteins and forms stable associations with TGF-BetaRI. It interferes with the phosphorylation of SMAD2 and the subsequent heteromerization with SMAD4, but does not inhibit the activity of SMAD3. The specificity of receptor–SMAD interactions is dictated by discrete structural elements in the receptor kinase domain and the MAD homology domain of the SMAD. Prior to activation, receptor regulated SMADs are anchored to the cell membrane by factors like SARA (SMAD Anchor for Receptor Activation) that brings the SMADs into proximity of the TGF receptor kinases (Ref.4).
TGF-Beta also induces other non-SMAD signaling pathways, which include activation of several MKKs (MAP kinase Kinase) and MEKs (MAPK/ERK Kinase) pathways (JNK/SPAK, p38, and ERK1/2) through upstream mediators RhoA, Ras, TAK1 (TGF-Beta Activated Kinase), TAB1 (TAK1 Binding Protein) and the proteins XIAP (Xenopus Inhibitor of Apoptosis), HPK1 (Haematopoietic Progenitor Kinase-1) are also involved in this link (Ref.2). Because of its critical role in cell fate determination, TGF-Beta signaling is subject to many levels of positive and negative regulation, targeting both the receptors and the intracellular mediators. Among the negative regulators of SMAD function are two highly conserved members of the Ski family of proto-oncoproteins c-Ski and c-SnON that antagonizes TGF-Beta signaling through direct interactions with the SMAD2/SMAD3 and SMAD4 (Ref.6) and later degrade releasing SMADs to regulate transcription. SMAD3 recruits the APC (Anaphase-Promoting Complex) and Cdh1 (Cadherin-1) to SnON, thus providing an alternative mechanism to target SnON for ubiquitination and degradation (Ref.3). Besides the direct induction or repression of target gene expression, TGF-Beta also induces a variety of complex cellular responses, depending on the cell type, most notably growth arrest in late G1 involving PI3K (Phosphatidylinositol-3-Kinase) and PP2A (Protein Phosphatase-2A), changes in differentiation programs, and apoptosis (Ref.3). Other growth factors also regulate TGF-Beta mediated signaling through GRB2 (Growth Factor Receptor-Bound Protein-2)-SOS activation of Ras.
TGF-Beta are possibly the most pleiotropic secreted proteins functioning as morphogens mediating several physiological processes including hematopoiesis, regulation of hormone secretion, in immune response, angiogenesis, tissue morphogenesis and regeneration, and bone induction and modulation. With respect to bone induction, TGF-Beta induces substantial endochondral bone formation in a muscle tissue site but limited bone formation in a bony site (Ref.1). Failure or dysregulation of TGF-Beta signaling is involved in the development of several diseases, such as hematological malignancies, i.e., Leukemia, Haemorragic Telangiectasia, Chondrodysplasias, impaired wound healing, neurodegenerative conditions, developmental disorders and Pulmonary Hypertension. Genetic or epigenetic loss of TGF-Beta signaling promotes tumorigenesis via suppression of the immune system and changes in cell differentiation of epithelial tumor cells, a phenomenon termed EMT (Epithelial Mesenchymal Transdifferentiation) (Ref.2). In addition, recent work has implicated TGF-Beta in other processes involved in tumor inhibition including maintenance of genomic stability, induction of senescence, suppression of telomerase activity and prevention of inappropriate angiogenesis. More recently in certain patients infected with HIV1 (Human Immunodeficiency Virus Type-1), increased levels of TGF-Beta promoted the production of virus and also impaired the host immune system suggesting a possible role in HIV1 viral gene regulation and pathogenesis (Ref.7).
References
- 1
- Ripamonti U, Crooks J, Matsaba T, Tasker J. Induction of endochondral bone formation by recombinant human transforming growth factor-beta2 in the baboon (Papio ursinus).
- 2
- Moustakas A, Pardali K, Gaal A, Heldin CH. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation.
- 3
- Roberts AB, Derynck R. Meeting report: signaling schemes for TGF-beta.
- 4
- Imai Y, Kurokawa M, Izutsu K, Hangaishi A, Maki K, Ogawa S, Chiba S, Mitani K, Hirai H. Mutations of the SMAD4 gene in acute myelogeneous leukemia and their functional implications in leukemogenesis.
- 5
- Huo YY, Zhang KT, Li BY, Duan RF, Fan BX, Xiang XQ, Hu YC, Xie L, Wu DC. Regulation of SMAD7 gene by TGF-beta 1 in process of malignant transformation.
- 6
- Wu JW, Krawitz AR, Chai J, Li W, Zhang F, Luo K, Shi Y. Structural mechanism of SMAD4 recognition by the nuclear oncoprotein Ski: insights on Ski-mediated repression of TGF-beta signaling.
- 7
- Li JM, Shen X, Hu PP, Wang XF. Transforming growth factor beta stimulates the human immunodeficiency virus 1 enhancer and requires NF-kappaB activity.
 关于我们
关于我们