PTEN_Pathway
发布时间:2019-12-11 11:25 来源:SABiosciences
- 通路
- 概述
Review
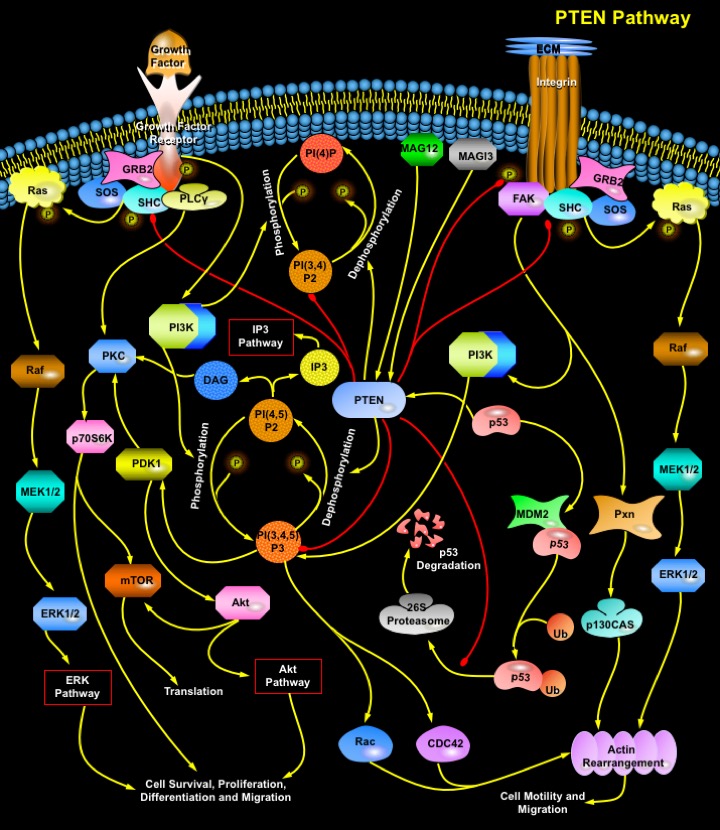
Tumorigenesis is the result of abnormal activation of growth programs in the cells. Cancer cells escape normal growth control mechanisms as a consequence of activating mutations, or increased expression of one or more cellular protooncogenes, and/or inactivating mutations, or decreased expression of one or more tumor suppressor genes. Most oncogene and tumor suppressor gene products are components of signal transduction pathways that control cell cycle entry or exit, promote differentiation, sense DNA damage and initiate repair mechanisms, or regulate cell death programs. PTEN (Phosphatase and Tensin Homologue Deleted from Chromosome-10)/MMAC1 (Mutated in Multiple Advanced Cancers-1)/TEP1 (TGF-Beta Regulated and Epithelial Cell Enriched Phosphatase-1) has recently been identified as one of the most frequently mutated tumor suppressors in human cancer that functions primarily as a cytoplasmic phosphatase to regulate crucial signal transduction pathways involving growth, adhesion, migration, invasion and apoptosis (Ref.1). It has been demonstrated to be involved in the regulation of a variety of phenotypes whereas its major function of tumour suppression is achieved by the downregulation of the oncogenic Akt/PKB (Protein Kinase-B) pathway (Ref.2). PTEN is expressed throughout the embryo, and has critically important functions in early embryonic development (Ref.3).
PTEN functions both as a dual specificity protein phosphatase and an inositol phospholipid phosphatase. Although it can dephosphorylate protein substrates such as FAK (Focal Adhesion Kinase) and the adapter protein SHC, PTEN's predominant enzymatic activity appears to be the dephosphorylation of phosphoinositides: PI(3,4,5)P3 (Phosphatidylinositol 3,4,5-trisphosphate) and PI(3,4)P2 (Phosphatidylinositol 3,4-bisphosphate) at the D3 positions into PI(4,5)P2 (Phosphatidylinositol 4,5-bisphosphate) and PI(4)P (Phosphatidyl Inositol Phosphate) respectively, thus antagonizing the PI3K (Phosphatidylinositiol–3 Kinase) activation (Ref.4). PTEN consists of a phosphatase domain that has a structure resembling protein tyrosine phosphatases but containing an enlarged active site that accounts for its ability to bind its PI (Phosphatidyl Inositol) substrates. There is a second major domain that binds phospholipids. This C2 domain appears to bind PTEN to the plasma membrane, and it might orient the catalytic domain appropriately for interactions with phosphatidylinositols and other potential substrates (Ref.5). Mutagenesis studies demonstrate the C-terminal tail of PTEN to be intriguing regulatory features that induce phosphorylation of certain serine and threonine residues (S380, T382 and T383) and can modulate both the enzymatic activity and the stability of PTEN. Loss of PTEN function, either in murine ES (Embryonic Stem) cells or in human cancer cell lines, results in accumulation of PI(3,4,5)P3 and PI(3,4)P2 and the subsequent activation of its downstream effectors, such as PDK-1 (Phosphoinositide-Dependent Kiinase-1), Akt, Btk family tyrosine kinases, PKC (Protein Kinase-C), and GEFs (Guanine Nucleotide Exchange Factors) for the Rho family of small GTPases (Guanosine Triphosphatases): Rac1 and CDC42 (Ref.4). PTEN is capable of inducing apoptosis or a cell cycle arrest, and loss of PTEN in primary cells leads to either excessive proliferation or defects in apoptosis (Ref.6).
The PI3K-->Akt pathway is a well-known oncogenic signaling pathway activated in response to various Growth Factors and ECM (Extracellular Matrix) proteins. Extracellular interactions trigger signaling from Integrins and Growth Factor Receptors and PI3K is activated. Activated PI3K brings about phosphorylation of phosphoinositides: PI(4,5)P2 (Phosphatidylinositol 4,5-bisphosphate) and PI(4)P (Phosphatidyl Inositol Phosphate) into PI(3,4,5)P3 (Phosphatidylinositol 3,4,5-trisphosphate) and PI(3,4)P2 (Phosphatidylinositol 3,4-bisphosphate) respectively. PI(3,4,5)P3 catalyzes the phosphorylation of Akt by PDK-1 at Thr308 within the activation loop and at Ser473 near the carboxyl terminus. On the other hand, accumulation of PI(4,5)P2 by PTEN leads to the activation of the molecular messengers: IP3 (Inositol Triphosphate) and DAG (Diacylglycerol). Activated Akt regulates complex downstream pathways affecting cell growth, survival and migration. It prevents cells from undergoing apoptosis by inhibiting the proapoptotic factors BAD and the cell death pathway enzyme Caspase9 as well as the nuclear translocation of FKHR and FKHRL1. In its active state FKHR, can promote a G1 arrest through the induction of p27 and can induce apoptosis perhaps through regulation of Fas signaling or through regulation of FasL (Fas Ligand) itself. In addition to promoting cell survival, Akt regulates cell proliferation and cell cycle progression by means of the phosphorylation of other targets: inhibition of GSK3 that contributes to Cyclin-D accumulation and cell cycle entry, activation of p70S6K (p70S6 Kinase) that contributes to cell growth by regulating translation of key mRNAs and down-regulation of p27, a major inhibitor for G1 Cyclin-dependent kinases (Ref.1). Recent evidence places the mTOR (Mammalian Target of Rapamycin) kinase downstream of the PI3K-->Akt signaling pathway, which is upregulated in multiple cancers because of loss of the PTEN. PTEN acts as an antagonist of cell survival by downregulating the Akt pathway through dephosphorylation and thus, inhibition of PI(3,4,5)P3. Maintenance of the PTEN tumor suppressor protein is required to modulate Akt activity and to concomitantly control the transcriptional activity of the anti-apoptotic transcription factor NF-KappaB (Nuclear Factor-KappaB) (Ref.7, 8, 9 & 10).
PTEN suppresses migration of a variety of cell types, including primary human fibroblasts and tumor cells primarily due to its ability to dephosphorylate proteins rather than lipids. The regulation of cell migration and adhesion can be divided into two components: (1) a directionally persistent migratory component promoted by the FAK-->Pxn (Paxillin)-->p130CAS signaling pathway, which involves enhanced orientation of the actin cytoskeleton, increased focal contacts and directional migration; and (2) a random-motility component promoted by association of SHC with GRB2 (Growth Factor Receptor-bound Protein-2) and subsequent activation of the Ras-->Raf-->MEK1/2-->ERK1/2 pathway, which involves a modest increase in F-actin levels, random orientation of the actin cytoskeleton and more random cell movement (Ref.3). The FAK signaling pathway is activated by Integrin receptors and is linked to cell motility and migration and other cellular activities like cell survival, proliferation and differentiation. The SHC-GRB2 pathway is activated by receptors that include various tyrosine kinase receptors and Integrins (Ref.5). PTEN's protein phosphatase activity is able to down-regulate both the pathways by direct dephosphorylation of FAK and SHC which leads to the inactivation of the Ras-->MAPK pathway. PTEN-regulated FAK and SHC activity further appears to impinge on cell adhesion, cell migration, and cell invasion. It therefore emerges that the loss of PTEN activity may confer increased survival ability, proliferative potential, and invasive capacity on cells, and thereby may promote progression towards a more malignant phenotype (Ref.5, 7 & 11).
PTEN expression and function is regulated through the action of various proteins. PTEN is able to autoregulate its expression and function through the stabilization of another tumor suppressor p53. PTEN enhances p53 transactivation, a relationship that requires the interaction between PTEN and p53 and is PTEN phosphatase independent. A network involving the mutual dependence of PTEN and p53 has emerged. Specifically, PTEN may protect p53 from MDM2-mediated degradation, whereas p53 can enhance the transcription of PTEN. MDM2 binds to the transactivation domain of p53, precluding interaction with the transcriptional machinery. This binding mediates the covalent attachment of Ub (Ubiquitin) to p53. Ubiquitylated p53 is then transported from the nucleus into the cytoplasm, where it is degraded by the Proteasome. PTEN acts to protect p53 from MDM2-mediated degradation by the Proteasome (Ref.11 &12). PTEN activity is also enhanced by the membrane-associated guanylate kinase family proteins with multiple PDZ domains: MAGI2 (Membrane Associated Guanylate Kinase, WW and PDZ Domain Containing-2) and MAGI3 (Membrane Associated Guanylate Kinase, WW and PDZ Domain Containing-3). PTEN binds to the PDZ domains MAGI2 and MAGI3 through an interaction between the PDZ-binding motif of PTEN and the second PDZ domain of MAGI-2. Thus, Both MAGI2 and MAGI3 enhance the ability of PTEN to suppress activation FAK, SHC and Akt, thus downregulating cell growth, survival and migration (Ref 13 & 14).
The PTEN tumour suppressor is among the most commonly inactivated or mutated genes in human cancer. Macroautophagy, a multistep process responsible for the degradation of long-lived proteins and organelle renewal, is regulated by PTEN and has been shown to be associated with human pathologies, including some forms of cardiomyopathy (Danon's disease) and breast cancer. Discovered in 1997 as a tumor suppressor gene located on human chromosome 10q23, PTEN has since been the focus of particularly intense interest because of its central role in suppressing tumours. Mutations in the gene are frequent in glioblastomas, endometrial carcinoma, melanomas, and prostate cancer. Furthermore, three dominant inherited disorders: Cowden disease, Lhermitte-Duclos disease and Bannayan-Zonana syndrome, are linked to germ line mutations in PTEN (Ref.4). Patients with these disorders develop widespread benign tumors (harmartomas) in the skin, mouth, thyroid, breast, intestine, and other tissues. They also have an increased risk of breast and thyroid cancer, cerebellar tumors, and mental retardation (Ref.3). Elucidation of the PTEN pathway stands as a potentially important advance in molecular oncology and the tumor status of PTEN seems to be of utmost importance in determining and monitoring treatment of an array of malignancies when the PTEN pathway has to be targeted for therapeutic intervention using new and/or existing pharmacological agents (Ref.15).
References
- 1
- Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis.
- 2
- Steelman LS, Bertrand FE, McCubrey JA. The complexity of PTEN: mutation, marker and potential target for therapeutic intervention.
- 3
- Wechsler-Reya R, Scott MP. The developmental biology of brain tumors.
- 4
- Stocker H, Andjelkovic M, Oldham S, Laffargue M, Wymann MP, Hemmings BA, Hafen E. Living with lethal PIP3 levels: viability of flies lacking PTEN restored by a PH domain mutation in Akt/PKB.
- 5
- Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis.
- 6
- Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN.
- 7
- Wu H, Goel V, Haluska FG. PTEN signaling pathways in melanoma.
- 8
- Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR.
- 9
- Tsutsui S, Inoue H, Yasuda K, Suzuki K, Tahara K, Higashi H, Era S, Mori M. Inactivation of PTEN is associated with a low p27Kip1 protein expression in breast carcinoma.
- 10
- Mayo MW, Madrid LV, Westerheide SD, Jones DR, Yuan XJ, Baldwin AS Jr, Whang YE. PTEN blocks tumor necrosis factor-induced NF-kappa B-dependent transcription by inhibiting the transactivation potential of the p65 subunit.
 关于我们
关于我们