NFKappaB_Family_Pathway
发布时间:2019-12-11 10:50 来源:SABiosciences
- 通路
- 概述
Review
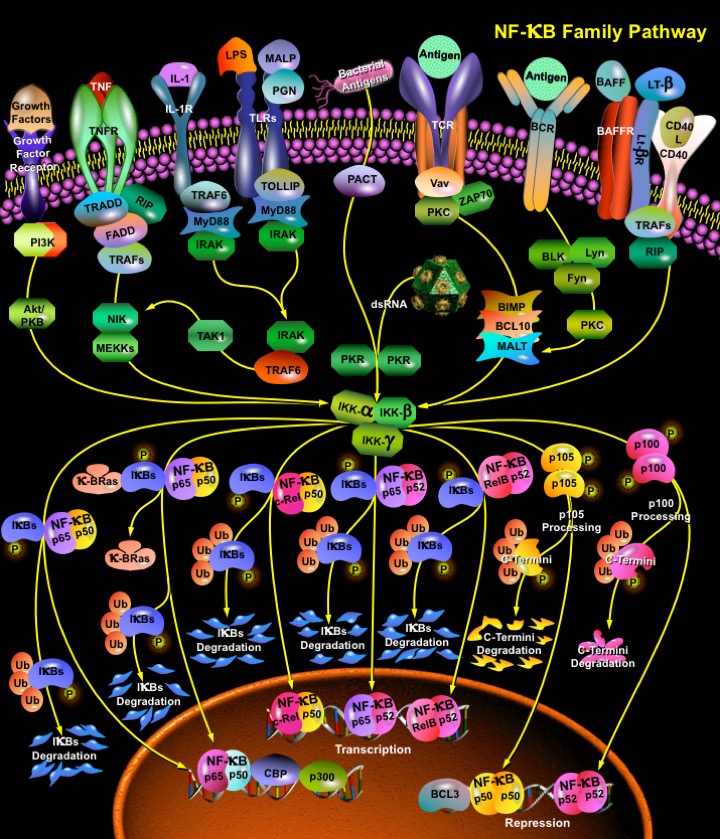
NF-KappaB (Nuclear Factor-KappaB) is a heterodimeric protein composed of different combinations of members of the Rel family of transcription factors. The Rel/ NF-KappaB family of transcription factors are involved mainly in stress-induced, immune, and inflammatory responses. In addition, these molecules play important roles during the development of certain hemopoietic cells, keratinocytes, and lymphoid organ structures. More recently, NF-KappaB family members have been implicated in neoplastic progression and the formation of neuronal synapses. NF-KappaB is also an important regulator in cell fate decisions, such as programmed cell death and proliferation control, and is critical in tumorigenesis (Ref.1).
NF-KappaB is composed of homo- and heterodimers of five members of the Rel family including NF-KappaB1(p50), NF-KappaB2 (p52), RelA (p65), RelB, and c-Rel (Rel). Hetero and Homo-dimerization of NF-KappaB proteins which exhibit differential binding specificities includes p50/RelA, p50/c-Rel, p52/c-Rel, p65/c-Rel, RelA/RelA, p50/p50, p52/p52, RelB/p50 and RelB/p52. All the Rel proteins contain a conserved N-terminal region, called the RHD (Rel Homology Domain). The N-terminal part of the RHD contains the DNA-binding domain, whereas the dimerization domain is located in the C-terminal region of the RHD. Close to the C-terminal end of the RHD lies the NLS (Nuclear Localization Signal), which is essential for the transport of active NF-KappaB complexes into the nucleus. NF-KappaB1 and RelA were the first NF-KappaB proteins to be identified. Their N-terminal 300 AA revealed high similarity to the oncoprotein v-Rel, its cellular homologue c-Rel and the Drosophila protein Dorsal what resulted in the terms Rel proteins and RHD. The Rel/ NF-KappaB proteins can be divided into two groups: Only RelA (p65), RelB and c-Rel (and Dorsal and Dif in Drosophila) contain potent TDs (Transactivation Domains) within sequences C-terminal to the RHD. The TDs consist of abundant serine, acidic and hydrophobic aminoacids that are essential for transactivation activity. In contrast, p50 and p52 do not possess TDs, and therefore cannot act as transcriptional activators by themselves. NF-KappaB1and NF-KappaB2 are produced as p105 and p100 precursors, respectively. The NF-KappaB1 p105 precursor appears to undergo constitutive processing by the cellular proteasome that removes the C-terminal I-KappaB-like portion to generate p50. NF-KappaB2 p100 precursor can be processed to remove the I-KappaB-like C-terminus, allowing the active p52 N-terminal half to function in transcriptional regulation. Homo- or heterodimers of p50 and p52 were even reported to repress KappaB site-dependent transcription, possibly by competing with other transcriptionally active dimers (e.g. p50/RelA) for DNA binding (Ref.2).
NF-KappaB dimers are sequestered in the cytosol of unstimulated cells via non-covalent interactions with a class of inhibitor proteins, called I-KappaBs. To date seven I-KappaBs have been identified: I-KappaB-alpha, I-KappaB-beta, I-KappaB-gamma, I-KappaB-epsilon, BCL3, p100 and p105. All known I-KappaBs contain multiple copies of a 30-33 aa sequence, called ankyrin repeats which mediate the association between I-KappaB and NF-KappaB dimers. The ankyrin repeats interact with a region in the RHD of the NF-KappaB proteins and by this mask their NLS and prevent nuclear translocation. Signals that induce NF-KappaB activity cause the phosphorylation of I-KappaBs, their dissociation and subsequent degradation, thereby allowing activation of the NF-KappaB complex. Activated NF-KappaB complex translocates into the nucleus and binds DNA at Kappa-B-binding motifs such as 5-prime GGGRNNYYCC 3-prime or 5-prime HGGARNYYCC 3-prime (where H is A, C, or T; R is an A or G purine; and Y is a C or T pyrimidine) and induce gene expression. The degradation of I-KappaB proteins that permits NF-KappaB molecules to move into the nucleus is also carried out by the proteasome but only after prior phosphorylation of I-KappaB by the IKK (I-KappaB Kinase Complex). The IKK is composed of three subunits: two, IKK-alpha (IKK1) and IKK-beta (IKK2), are bonafide kinases, while the third, IKK-gamma (NEMO), has no catalytic activity but plays a critical regulatory role. IKK-alpha is the predominant I-KappaB kinase. Phosphorylated I-KappaB is recognized by Beta-TrCP, a component of the SCF (skp-1/ Cul/F box) ubiquitin ligase complex that mediates poly-ubiquitination of I-KappaB and its subsequent proteasomal degradation. In contrast, IKK-alpha mediates the phosphorylation-dependent processing of p100, resulting in the generation of p52 (Ref.3).
NF-KappaB can be activated by exposure of cells to LPS (Lipopolysaccharides) or inflammatory cytokines such as TNF (Tumour Necrosis Factor) or IL-1 (Interleukin-1), growth factors, lymphokines, oxidant-free radicals, inhaled particles, viral infection or expression of certain viral or bacterial gene products, UV irradiation, B or T-Cell activation, and by other physiological and non physiological stimuli. The most potent NF-KappaB activators are the proinflammatory cytokines IL-1 and TNF, which cause rapid phosphorylation of KappaBs at two sites within their N-terminal regulatory domain. TNF, which is the best-studied activator, binds to its receptor and recruits a protein called TRADD (TNF-Associated Receptor Death Domain). TRADD binds to the TRAF2 (TNF Receptor-Associated Factor-2) that recruits NIK (NF-KappaB-Inducible Kinase). Both IKK1 and IKK2 have canonical sequences that can be phosphorylated by the MAP (Mitogen Activated Protein) kinase NIK/MEKK1 and both kinases can independently phosphorylate I-KappaB-alpha or I-KappaB-beta. TRAF2 also interacts with A20, a zinc finger protein whose expression is induced by agents that activate NF-KappaB. A20 functions to block TRAF2-mediated NF-KappaB activation. A20 also inhibits TNF and IL-1 induced activation of NF-KappaB suggesting that it may act as a general inhibitor of NF-KappaB activation. CD40, another member of the TNF receptor family, can signal the induced processing of p100 to p52. The ligand for CD40, CD40L (CD154), is expressed on activated CD4-T cells, and when it engages CD40 in a T:B interaction, can induce B-Cell proliferation and differentiation. CD40 signaling induces p100 processing through NIK in the non-canonical pathway. LT-BetaR (Lymphotoxin–Beta Receptor), which is critically important for the development and organization of lymphoid tissue also gives way to two separate pathways, one that activates the canonical NF-KappaB pathway and depends upon IKK-beta and IKK-gamma/NEMO and another that induces p100 processing dependent on NIK and IKK-alpha (Ref.4).
The recognition of bacterial and viral products by Toll-like receptors on cells of the innate immune system also results in NF-KappaB induction, leading to the production of proinflammatory cytokines and the activation of Antigen Presenting Cell for T-Cell costimulation in the adaptive immune response. Viral infection leads to the increased expression and secretion of the cytokine interferon gamma (IFN-gamma) from host cells. IFN-gamma activates the double-stranded RNA (dsRNA)-dependent serine-threonine protein kinase R (PKR). dsRNA produced during viral replication induces PKR dimerization, autophosphorylation, and activation of the eIF-2alpha kinase activity. When eIF-2alpha is phosphorylated, cellular and viral protein translation cannot efficiently occur. Alternatively, bacterial products or cellular stress can also activate PKR by an endogenous gene product called PACT. The binding of PACT to PKR promotes conformational changes that allow PKR to activate the downstream signaling pathways leading to the activation of NF-KappaB. Several survival and stimulatory factors that is important in the development and BAFF (B-Cell activating factor) coordinated response of B and T lymphocytes also employ NF-KappaB to carry out their instructive functions. BAFF induces B-Cell survival and development and requires the specific BAFF receptor, BAFFR (BR3), as well as NF-KappaB2, and the NIK kinase. In both immature and mature B cells, BAFF induced processing of p100 to p52, dependent on BAFF-R and NIK, and in addition this process is independent of the canonical IKK complex (Ref.5).
The interaction of the Antigen Presenting Cell and T-Cell also causes NF-KappaB activation in both cell types NF-KappaB activation is triggered in T-Cells by the engagement of the T cell receptor and the CD28 receptor with their ligands, MHC class II, and the costimulatory molecules CD80 and CD86 presented by Antigen Presenting Cells. The T-Cell receptor and CD28 synergize in induction of the NF-KappaB -dependent genes required for T-Cell activation and proliferation, such as IL-2, IL-2 receptor (IL-2R), and IFN (Interferons). Activated T-Cells, in turn, elicit NF-KappaB activation in Antigen Presenting Cells (Ref.6).
Exposure of cells to different forms of radiation and other genotoxic stresses stimulates signaling pathways that activate transcription factor NF-KappaB, which elicit various biological responses through induction of target genes. UV-C or UV-B also induces NF-KappaB activity. In addition to short wavelength UV radiation, NF-KappaB activity is also induced by exposure to even shorter wavelength photons, gamma rays or IR (Ionizing Radiation). Although both radiations induce I-KappaB degradation they operate through two distinct mechanisms. Whereas IR exposure results in IKK activation and IR-induced I-KappaB degradation is phosphorylation dependent, exposure to UV-C does not result in IKK activation, and UV-induced I-KappaB degradation occurs independently of its N-terminal serine phosphorylation (Ref.7).
Several Growth factors also activates NF-KappaB. HGF (Hepatocyte Growth Factor) stimulation enhances both NF-KappaB DNA binding and NF-KappaB-dependent transcriptional activity. The signaling mechanisms mediating these effects include the classical I-KappaB-alpha phosphorylation-degradation cycle, as well as the ERK1/2 (Extracellular Signal-Regulated Kinases-1 and -2) and p38 MAPK. NF-KappaB activation contributes to HGF-mediated proliferation and tubulogenesis. Another pathway, which has been implicated in the enhancement of NF-KappaB-dependent transcription, is PI3K/Akt. An essential role for PI3K/Akt in enhancing the transcriptional activity of the NF-KappaB p65 subunit has been described downstream of TNF-Alpha or IL-1. However, this pathway does not seem to be required in all cell types or for all stimuli (Ref.8).
NF-KappaB was also found to stimulate transcription of Cyclin-D1, a key regulator of G1 checkpoint control. Two NF-KappaB binding sites in the human Cyclin-D1 promoter conferred activation by NF-KappaB as well as by growth factors. Both levels and kinetics of Cyclin-D1 expression during G1 phase were controlled by NF-KappaB. Moreover, inhibition of NF-KappaB caused a pronounced reduction of serum-induced Cyclin-D1 associated kinase activity and resulted in delayed phosphorylation of the retinoblastoma protein. Inappropriate activation of NF-KappaB has been linked to inflammatory events associated with autoimmune arthritis, asthma, septic shock, lung fibrosis, glomerulonephritis, atherosclerosis, and AIDS. In contrast, complete and persistent inhibition of NF-KappaB has been linked directly to apoptosis, inappropriate immune cell development, and delayed cell growth (Ref.9).
References
- 1
- Baldwin AS Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights.
- 2
- Vigo Heissmeyer, Daniel Krappmann, Eunice N. Hatada, and Claus Scheidereit. Shared Pathways of I B Kinase-Induced SCF TrCP-Mediated Ubiquitination and Degradation for the NF- B Precursor p105 and I B .
- 3
- Baeuerle, P. A. I-kappa-B--NF-kappa-B structures: at the interface of inflammation control.
- 4
- Joel L. Pomerantz and David Baltimore. Two pathways to NF-KappaB.
- 5
- Fulvio D\'Acquisto, and Sankar Ghosh. PACT and PKR: Turning on NF-KappaB in the Absence of Virus.
- 6
- Vishva Dixit and Tak. W. Mak. NF-KappaB signaling: Many Roads Lead To Madrid.
- 7
- Nanxin Li and Michael Karin. Ionizing radiation and short wavelength UV activate NF-KappaB through two distinct mechanisms.
- 8
- Markus Müller, Alessandro Morotti, and Carola Ponzetto. Activation of NF- B Is Essential for Hepatocyte Growth Factor-Mediated Proliferation and Tubulogenesis.
- 9
- Michael Hinz, Daniel Krappmann, Alexandra Eichten, Andreas Heder, Claus Scheidereit, and Michael Strauss. NF-KappaB Function in Growth Control: Regulation of Cyclin D1 Expression and G0/G1-to-S-Phase Transition.
 关于我们
关于我们