NFAT_Cardiac_Hypertrophy
发布时间:2019-12-11 10:42 来源:SABiosciences
- 通路
- 概述
Review
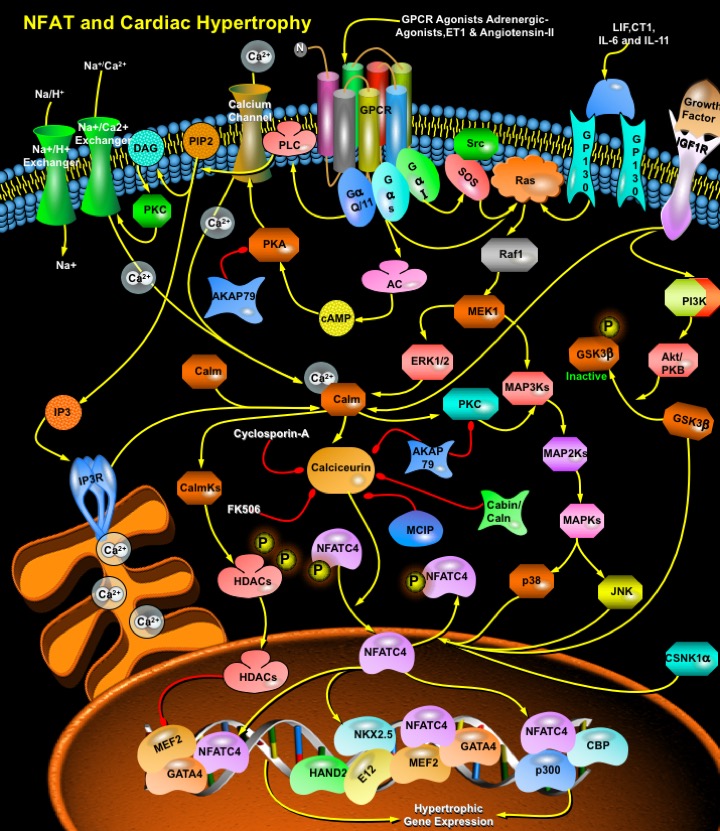
Cardiac failure, one of the largest health care burdens in the United States and other developed countries is often associated with prolonged and maladaptive cardiac hypertrophy, defined as a compensatory mechanism of the heart that helps to maintain cardiac output during pathological states with sustained increases in hemodynamic load (Ref.1). As cardiomyocytes lose the ability to divide soon after birth, cardiac hypertrophy offers an important adaptive response in vivo that allows the organism to maintain or increase its cardiac output. The adult myocardium responds to a wide array of intrinsic and extrinsic stimuli, including hypertension, myocardial infarction, cardiac arrhythmias, valvular disease, endocrine disorders, increased workload, injury, and contractile abnormalities resulting from mutant sarcomeric proteins by hypertrophic growth (Ref.2). Although initially beneficial, prolonged hypertrophy is correlated with poor clinical prognosis resulting in dilated cardiomyopathy, heart failure, and sudden death (Ref.3).
The hypertrophic response is orchestrated by growth factors and cytokines acting through several interdependent signaling cascades, including, specific G-protein isoforms, low-molecular-weight GTPases (Ras, RhoA, and Rac), MAPK (Mitogen-Activated Protein Kinase) and PKC (Protein Kinase-C) (Ref.1). In addition, recent studies have recognized the importance of Ca2+-sensitive signaling molecules, including calcineurin, CalmK (Calcium/Calmodulin-Dependent Protein Kinase), and MAPK in hypertrophic pathways, in which NFAT (Nuclear Factor Of Activated T-Cells) occupies the central place, being the best-characterized target for the development of cardiac hypertrophy (Ref.4). NFAT is a family of transcription factors composed of four structurally related members: NFATc, NFATp, NFAT3, and NFAT4, also denominated, NFAT2/NFATc1, NFAT1/NFATc2, NFATc4, and NFATc3 respectively, that are expressed in the cytoplasm of the resting cells, as well as the constitutively nuclear NFAT5 member (Ref.2), out of which, NFAT3/ NFATc4 is expressed in the adult heart and mediates the signals of cardiac hypertrophy (Ref.5). Initial phase in the development of myocardial hypertrophy involves the formation of cardiac para- and/or autocrine factors like Endothelin-1, Angiotensin-II, and the Adrenergic agonists: phenylephrine and norepinephrine, at the cell membrane, the receptors of which are coupled to G-proteins of the G-AlphaQ/11, G-AlphaS and G-AlphaI-families. Other receptors associated with the induction hypertrophy through the NFATc4 pathway include IGF1 (Insulin Like Growth Factor-1), GP130 etc.
GPCRs (G-Protein Coupled Receptors) play an important role in the regulation of cardiac function and adaptation to changes in hemodynamic burden. These heptahelical receptors are coupled to three principal classes of heterotrimeric GTP-binding proteins, G-AlphaS, G-AlphaQ/11, and G-AlphaI, which transduce the agonist or antagonist-induced signals towards intracellular effectors such as enzymes and ion channels. All G-Proteins consist of the subunits G-Alpha and G-BetaGamma, which upon receptor activation dissociate and independently activate intracellular signaling pathways. Angiotensin-II, Endothelin-1, and Alpha-Adrenergic receptors are coupled to G-AlphaQ/11 that in turn activates PLC-Gamma (Phospholipase-C-Gamma). Activation of PLC-Gamma leads to increased hydrolysis of membrane PIP2 (Phosphatidylinositol-4,5-Bisphosphate), the products of which are IP3 (Inositol Trisphosphate) and DAG (Diacylglycerol). DAG binds to and activates PKC that phosphorylates numerous substrates. Activation of PKC leads to changes in Ca2+ handling (through phosphorylation of channels and pumps) and increases in NHE1 (Na+/H+ exchanger-1) (Ref.6). IP3 binds to IP3R (IP3 Receptors) on the surface of the ER (Endoplasmic Reticulum) leading to release of Ca2+ ions. The increased Ca2+ levels then activate the protein phosphatase calcineurin by disrupting the inhibitory effects of Calm. Calcineurin activation leads to the dephosphorylation of NFATc4, allowing it to enter the nucleus, where it cooperates with other transcription factors to participate in the formation transcriptional regulatory complexes.
The G-AlphaS subunit of ADR-Beta1 (Beta1-Adrenergic Receptor)in cardiac tissues activates AC (Adenyl Cyclase) and cAMP (cyclic Adenosine-3’,5’ Monophosphate) formation. Subsequently, activation of cAMP-dependent PKA (Protein Kinase-A) leads to phosphorylation of a set of regulatory proteins involved in cardiac excitation-contraction coupling and energy metabolism, including activation of L-Type Ca2+ channels, the SERCA (Sarco- and Endoplasmic Reticulum Ca2+ ATPase) eventually resulting in positively chronotropic, inotropic, and lusitropic effects on the heart. The less abundant ADR-Beta2 can couple to both G-AlphaS and G-AlphaI, which provide an additional level of regulation. G-BetaGamma released from the G-AlphaI-coupled ADR-Beta activates MAPK in an Src and Ras-dependent pathway. AC can be activated by several receptor tyrosine kinases, such as the IGF1 receptor, as well as GPCRs, including Alpha and ADR-Beta2 that contribute to PI3K-dependent pathways in the activation of NFATc4. The small G-Protein implicated in cardiac hypertrophy, Ras is also placed upstream of calcineurin and NFATc4 activation in cardiac myocytes in a Raf1-MEK1-dependent pathway involving MAP2Ks and MAP3Ks (Ref.7). Induction of GP130, a promiscuous receptor for several cytokines, including IL-6, IL-11 (Interleukins), LIF (Leukemia Inhibitory Factor), and Cardiotrophin-1 leads to activation of MAPK pathway, which results in the induction of genes involved in hypertrophy and survival pathways.
Activation of calcineurin by the upstream effectors dephosphorylates NFATc4, which translocates into the nucleus to interact with other transcription factors to regulate transcription. NFAT proteins cooperate with other transcription factors such as GATA4, E12, CBP (CREB Binding Protein), p300, MEF2 (Mads Box Transcription Enhancer Factor-2) etc., leading to activation of transcription of genes (ANF [Atrial Natriuretic Factor], Alpha-actin, Beta-myosin, TNF-Alpha [Tumor Necrosis Factor-Alpha], Endothelin-1 etc) essential for cardiac development, and thus, hypertrophy. The expression of Myosin Heavy Chain isoforms from alpha to beta and from alpha-cardiac to alpha-skeletal actin is also a feature of the calcineurin-NFATc4 pathway. (Ref.2). The ADSS (Adenylosuccinate Synthetase) gene expression is the outcome of the activation of the enhancer region that contains binding sites for NKX2.5, GATA4, MEF2, E12, HAND1 (Heart And Neural Crest Derivatives Expressed-1), and HAND2 (Ref.8). Action of MEF2, an essential factor for the transcription of Endothelin-1, ANF, and Myosin Heavy Chains etc. is regulated by direct association with HDACs (Histone Deacetylases).
NFATc4 remains in phosphorylated condition under basal, unstimulated conditions. Dephosphorylation mediated by the calcineurin, promotes nuclear translocation and subsequent transcriptional activation of NFATc4 whereas, rephosphorylation of the dephosphorylated NFATc4 terminates NFATc4 activation. MAPKs (p38MAPK, JNK) and additional kinases (e.g., GSK3Beta, CSNK1-Alpha [Casein Kinase-1Alpha]) mediate rephosphorylation of NFATc4 and thus act as negative regulators of cardiac hypertrophy. GSK3Beta, a widely expressed kinase phosphorylates a series of serine/threonine residues in the N-terminal regulatory regions of NFATc4 proteins, thereby masking their nuclear import sequences and promoting translocation to the cytoplasm and transcriptional inactivation. Rephosphorylated NFATc4 is exported out of the nucleus, and NFATc4-mediated transcription is terminated (Ref.9). In addition, calineurin activity is attenuated by several endogenous calcineurin inhibitors, such as AKAP79 (A-Kinase Anchor Protein-79-KD), Cabin/Cain, and DSCR1 (Down Syndrome Critical Region Gene-1)/MCIP (Myocyte-Enriched Calcineurin-Interacting Protein). AKAP79 interacts with calcineurin as well as PKA and PKC. Immunosuppressive drugs, Cyclosporin-A and FK506 inhibit calcineurin thereby blocking NFATc4 nuclear localization that leads to attenuation of cardiac hypertrophy (Ref.10).
References
- 1
- Fiedler B, Lohmann SM, Smolenski A, Linnemuller S, Pieske B, Schroder F, Molkentin JD, Drexler H, Wollert KC. Inhibition of calcineurin-NFAT hypertrophy signaling by cGMP-dependent protein kinase type I in cardiac myocytes.
- 2
- Olson EN, Molkentin JD. Prevention of cardiac hypertrophy by calcineurin inhibition: hope or hype?
- 3
- Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo.
- 4
- Johnson EN, Lee YM, Sander TL, Rabkin E, Schoen FJ, Kaushal S, Bischoff J. NFATc1 mediates vascular endothelial growth factor-induced proliferation of human pulmonary valve endothelial cells.
- 5
- Gomez del Arco P, Martinez-Martinez S, Maldonado JL, Ortega-Perez I, Redondo JM. A role for the p38 MAP kinase pathway in the nuclear shuttling of NFATp.
- 6
- Sugden PH. Signaling in myocardial hypertrophy: life after calcineurin?
- 7
- Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly.
- 8
- Wen HY, Xia Y, Young ME, Taegtmeyer H, Kellems RE. The adenylosuccinate synthetase-1 gene is activated in the hypertrophied heart.
- 9
- Yang TT, Xiong Q, Enslen H, Davis RJ, Chow CW. Phosphorylation of NFATc4 by p38 mitogen-activated protein kinases.
- 10
- Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, Molkentin JD. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic gro
 关于我们
关于我们