mTOR_Pathway
发布时间:2019-12-11 10:36 来源:SABiosciences
- 通路
- 概述
Review
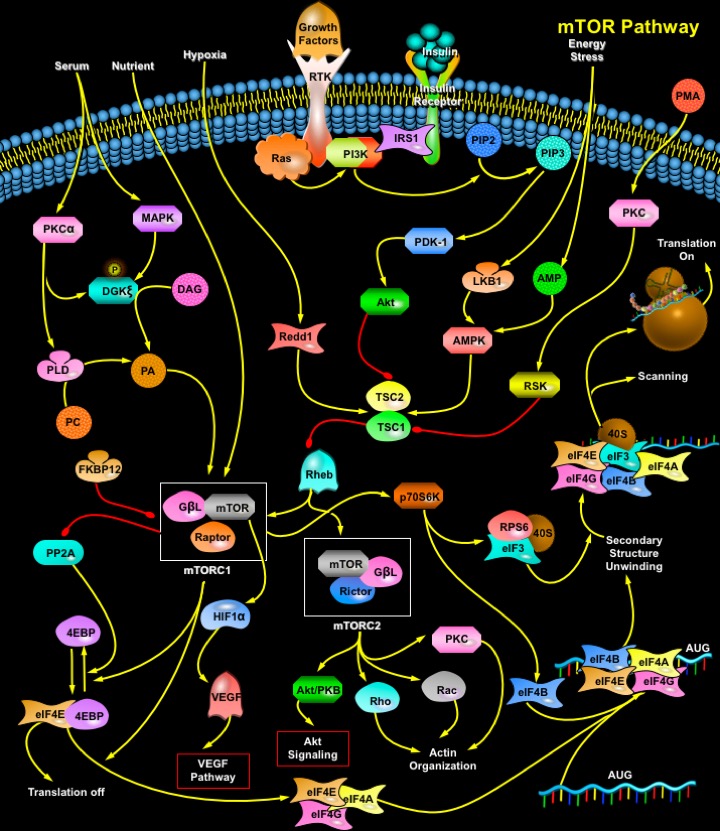
mTOR (Mammalian Target of Rapamycin) is a 289-kDa serine/threonine protein kinase and a member of the PIKK (Phosphatidylinositol 3-Kinase-related Kinase) family. The protein consists of a Catalytic Kinase domain, an FRB (FKBP12–Rapamycin Binding) domain, a putative Auto-inhibitory domain (Repressor domain) near the C-terminus and up to 20 tandemly repeated HEAT motifs at the Amino terminus, as well as FAT (FRAP-ATM-TRRAP) and FATC (FAT C-terminus) domains. The C-terminus of TOR is highly homologous to the catalytic domain of PI3K (Phosphatidylinositol 3-Kinase). TOR proteins are evolutionarily conserved from yeast to human in the C-domain, with human, mouse, and rat mTOR proteins sharing 95% identity at the amino acid level. The human mTOR gene encodes a protein of 2549 amino acids with 42% and 45% sequence identity to yeast TOR1 and TOR2, respectively. mTOR functions as a central element in a signaling pathway involved in the control of cell growth and proliferation (Ref.1).
The mTOR pathway is regulated by a wide variety of cellular signals, including Mitogenic Growth Factors, Hormones such as Insulin, Nutrients (Amino acids, Glucose), Cellular Energy Levels, and Stress conditions. A principal pathway that signals through mTOR is the PI3K/Akt (v-Akt Murine Thymoma Viral Oncogene Homolog-1) signal transduction pathway, which is critically involved in the mediation of cell survival and proliferation. Signaling through the PI3K/Akt pathway is initiated by mitogenic stimuli from Growth factors that bind receptors in the cell membrane. These receptors include IGFR (Insulin-like Growth Factor Receptor), PDGFR (Platelet-Derived Growth Factor Receptor), EGFR (Epidermal Growth Factor Receptor), and the Her family. The signal from the activated receptors is transferred directly to the PI3K/Akt pathway, or, alternatively, it can be activated through activated Growth Factor Receptors that signal through oncogenic Ras. Ras is another central switch for signal transduction and has been shown to be a pivotal activator of the MAPK (Mitogen-Activated Protein Kinase) signal transduction pathway. PI3K/Akt pathway can also be activated by Insulin via IRS1/2 (Insulin Receptor Substrate-1/2). Insulin binding activates the IR (Insulin Receptor) tyrosine kinase, which phosphorylates IRS1 or IRS2. PI3K binds phosphorylated IRS by SH2 (Src-Homology-2) domains in the p85 regulatory subunit. This interaction activates the p110 catalytic subunit. PI3K then catalyzes the conversion of membrane-bound PIP2 (Phosphatidylinositol (4,5)-bisphosphate) to PIP3 (Phosphatidylinositol (3,4,5)-triphosphate). PIP3 then binds the pleckstrin homology domain of Akt, which results in Akt activation through dimerization and exposure of its catalytic site. Akt can also be phosphorylated and activated by PDK-1 (Phospholipid-Dependent Kinase-1). Akt phosphorylates mTOR directly. Akt may also work indirectly on mTOR through the actions of the TSC1/TSC2 (Tuberous Sclerosis Complex). The physical association of the proteins TSC1 (Hamartin) and TSC2 (Tuberin) produces a functional complex that inhibits mTOR. Recent evidence indicates that the inhibitory effect of TSC1/TSC2 is mediated through TSC2 inactivation of a Ras family small GTPase known as RHEB (Ras Homolog Enriched in Brain). TSC2 has GAP (GTPase-Activating Protein) activity toward RHEB, and it has been postulated that the TSC1/TSC2 complex inhibits mTOR signaling by stimulating GTP hydrolysis of RHEB. RHEB-GTP activates mTOR. PMA (Phorbol Myristate Acetate) can also lead to mTOR phosphorylation independently of Akt through inhibition of the TSC1/2 complex via PKC (Protein Kinase-C) and RSK1 (Ribosomal-S6 Kinase-1) as well as through activation of S6K1 by PKC. AMPK (AMP (Adenosine 5'-Monophosphate)-Activated Protein Kinase) can also modulate mTOR. AMPK functions as the key energy-sensing kinase by virtue of its exquisite sensitivity to increases in the cellular AMP (Adenosine 5'-Monophosphate) /ATP (Adenosine Triphosphate) ratio. Increases in this ratio promote AMPK phosphorylation and activation by the upstream kinase LKB1, a human tumor suppressor mutated in Peutz-Jeghers syndrome. Activated AMPK in turn phosphorylates TSC2 (on residues distinct from those phosphorylated by Akt), apparently promoting its activation. This in turn inhibits the action of mTOR activity (Ref.2, 3 & 5).
PA (Phosphatidic Acid) can also activate mTOR. Three different enzymes generate PA: PLD (Phospholipase-D), LPAAT (Lysophosphatidic Acid Acyltransferase), and DGK (Diacylglycerol Kinase). PLD is regarded as the main contributor of PA to mTOR signaling. Nonetheless, other PA-generating enzymes can also contribute to mTOR activation; LPAAT is reported to be elevated in some tumors, and its overexpression leads to cell transformation. Serum stimulation leads to PLD activation, which correlates with increased mTOR signaling. Serum, a mixture of mitogenic agents, acts through GPCRs (G-Protein Coupled Receptors) or RTKs (Receptor Tyrosine Kinase). PLD activity increases in response to stimulation of both receptor types. Lipids such as DAG (Diacylglycerol) and PA are generated in membrane domains, where an intimate connection between distinct lipid metabolic pathways is maintained, to produce appropriate spatio-temporal responses. PLD and DGK may operate in parallel pathways, but they may also act as DAG- and PA-generating enzymes in a single pathway. In mammalian cells, the PA generated at internal membranes, such as Golgi, is produced mainly by PLD action on PC (Phosphatidylcholine). This PA can serve either as a messenger, promoting vesicle fission, or as a substrate for Phosphatases that transform PA to DAG. Because PC is the most abundant lipid in mammalian membranes, this pathway serves as a robust supplier of DAG that could then be used as a DGK substrate (Ref.4 & 5). Thus, several mechanisms have been proposed to explain how mTOR is regulated by growth factors and cellular energy levels. However, little is known as to how mTOR is regulated by stress conditions. Two stress-induced proteins, RTP801/Redd1 and RTP801L/Redd2, potently inhibit signaling through mTOR. RTP801 and RTP801L work downstream of AKT and upstream of TSC2 to inhibit mTOR functions. Another inhibitor of mTOR is Rapamycin. When complexed with its cellular receptor, FKBP12 (FK506 Binding Protein-12), Rapamycin binds directly to TOR to inhibit downstream signaling (Ref. 6 & 7).
Activation of mTOR results in phosphorylation of several downstream targets. For the protein mTOR to activate its signaling cascade, it must form the Ternary complex mTORC1 (mTOR Complex-1) and mTORC2 (mTOR Complex-2). Rapamycin-sensitive mTORC1 controls several pathways that collectively determine the mass (size) of the cell. Rapamycin-insensitive mTORC2 controls the actin cytoskeleton and thereby determines the shape of the cell. mTORC1 (and likely mTORC2) are multimeric, although are drawn as monomers. mTORC1 is a ternary complex containing mTOR, RAPTOR (Regulatory Associated Protein of mTOR) and G-BetaL (G-protein Beta-subunit-like protein). On the other hand mTORC2 complex consist of mTOR, G-BetaL and Rictor. The best-characterized effectors downstream of mTOR are 2 signaling pathways that act in parallel to control mRNA translation. Activated mTOR mediates the phosphorylation of the eIF4EBP1 (Eukaryotic Translation Initiation Factor-4E-Binding Protein-1) and the ribosomal protein p70S6K or S6K1 (S6 Kinase). 4EBP1 (also known as PHAS1 (Phosphorylated Heat-stable and Acid-Stable protein)) is a low molecular weight protein that can repress the activity of the eIF4F (eukaryotic Initiation Factor-4) complex. In its unphosphorylated state, 4EBP1/PHAS1 binds tightly to eIF4E (Eukaryotic Translation Initiation Factor-4E), the mRNA cap binding subunit of the eIF4F complex, which inhibits the activity of eIF4E in the initiation of protein synthesis. Phosphorylation of 4EBP1 by mTOR reduces its affinity for eIF4E, and the 2 proteins dissociate. eIF4E is then able to associate with the other components of eIF4F, which include the large scaffolding protein, eIF4G (Eukaryotic Translation Initiation Factor-4-Gamma), the adenosine triphosphate dependent RNA helicase eIF4A (Eukaryotic Translation Initiation Factor-4A), and eIF4B (Eukaryotic Translation Initiation Factor-4B), to form an active complex. This complex facilitates cap-dependent protein translation. The net effect is an increase in the translation of the subset of mRNAs with 5´ UTRs (Untranslated Regions), which often encode proteins associated with the proliferative response and the transition from G1 to S phase in the cell cycle. Such mRNAs include those that code for c-Myc, CcnD1 (Cyclin-D1), and Ornithine Decarboxylase. Cyclin-D1 binds with CDK4 (Cyclin-Dependent Kinase-4) to form a complex required for the phosphorylation of Rb (Retinoblastoma) protein, which subsequently contributes to progression of the cell cycle and DNA replication. Growth factor deprivation or inhibition of mTOR results in the dephosphorylation of 4EBP1, followed by its reassociation with eIF4E and a subsequent reduction in cap-specific translation. mTOR may also indirectly influence the phosphorylation state of 4EBP1 by modulating the activity of PP2A (Protein Phosphatase-2A). The second principal effector downstream of mTOR is the S6K1 serine/threonine kinase. After receiving a proliferative upstream signal mediated by the PI3K/Akt pathway, mTOR phosphorylates and activates S6K1. In turn, S6K1 phosphorylates and activates the 40S ribosomal S6 protein, facilitating the recruitment of the 40S ribosomal subunit into actively translating polysomes. In particular, the translation of mRNAs with 5´TOP (5´-Terminal Oligopyrimidine) sequence is enhanced. These 5´TOP mRNAs code primarily for ribosomal proteins, elongation factors and IGF-II (Insulin-like Growth Factor-II). Dephosphorylation of S6K1 decreases the synthesis of components of the protein translation system and results in a profound decrease in protein synthesis. mTORC1 also regulates VEGF (Vascular Endothelial Growth Factor) by phosphorylating HIF1Alpha (Hypoxia-Inducible Factor-1-Alpha Subunit) (Ref.8, 9 & 10).
In addition to its effects on translation, mTOR also modulates protein synthesis through regulation of RNA Polymerase I and III, which are responsible for the transcription of ribosomal and transfer RNAs. In the presence of appropriate Growth signals such as IGF1, mTOR, together with the PI3K and MAPK pathways, modulates Pol I-directed transcription of ribosomal RNAs. There is also evidence that mTOR may exert its effects on the polymerases through regulation of the phosphorylation status of Rb by influencing the stability and expression of Cyclin-D1 and p27, both of which regulate CDKs upstream of Rb. mTORC2 may signal to the actin cytoskeleton through a small Rho-type GTPase and PKC. Furthermore, mTORC2 controls the formation of activated, GTP-bound Rac1 in a growth-factor-dependent fashion. mTORC2 also controls the phosphorylation and activation of PKC-Alpha (Protein Kinase-C-Alpha). mTOR as a central modulator of proliferative signal transduction is an ideal therapeutic target against cancer. Through extensive clarification of many signal transduction pathways, it has become clear that the mTOR kinase participates in critical events that integrate external signals with internal signals, coordinating cellular growth and proliferation. mTOR receives signals that indicate whether transcription and translational machinery should be upregulated, then efficiently transmits these signals to the appropriate pathways. Multiple components of pathways that signal through mTOR are dysregulated in numerous cancer types. The development of inhibitors of mTOR is a rational therapeutic strategy for malignancies that are characterized by dysregulated pathways that signal through mTOR (Ref.9 & 11).
References
- 1
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism.
- 2
- O\'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt.
- 3
- Jozwiak J, Jozwiak S, Grzela T, Lazarczyk M. Positive and negative regulation of TSC2 activity and its effects on downstream effectors of the mTOR pathway.
- 4
- Avila-Flores A, Santos T, Rincon E, Merida I. Modulation of the mammalian target of rapamycin pathway by diacylglycerol kinase-produced phosphatidic acid.
- 5
- Han S, Khuri FR, Roman J. Fibronectin stimulates non-small cell lung carcinoma cell growth through activation of Akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways.
- 6
- Cortot A, Armand JP, Soria JC. PI3K-AKT-mTOR pathway inhibitors.
- 7
- Ellisen LW. Growth control under stress: mTOR regulation through the REDD1-TSC pathway.
- 8
- Fumarola C, La Monica S, Alfieri RR, Borra E, Guidotti GG. Cell size reduction induced by inhibition of the mTOR/S6K-signaling pathway protects Jurkat cells from apoptosis.
- 9
- Lang CH, Frost RA. Endotoxin disrupts the leucine-signaling pathway involving phosphorylation of mTOR, 4E-BP1, and S6K1 in skeletal muscle.
- 10
- Fiano V, Ghimenti C, Imarisio S, Silengo L, Schiffer D. PAkt, cyclin D1 and p27/Kip.1 in glioblastomas with and without EGFR amplification and PTEN mutation.
 关于我们
关于我们