JNK_Pathway
发布时间:2019-12-11 09:21 来源:SABiosciences
- 通路
- 概述
Review
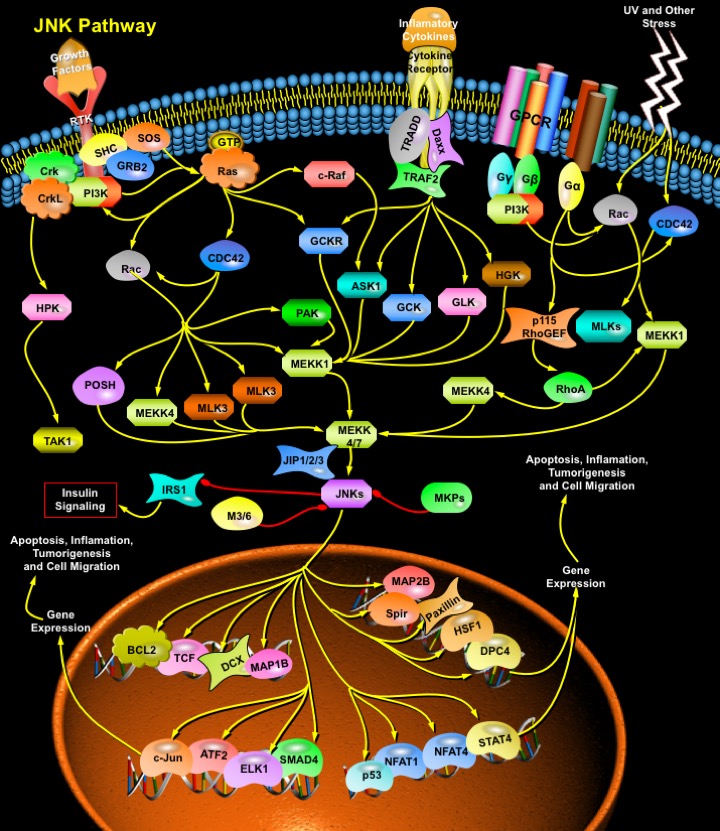
MAPKs (Mitogen-Activated Protein Kinases) are Serine-threonine protein Kinases that are activated in response to a variety of extracellular stimuli and mediate signal transduction from the cell surface to the nucleus. MAPKs are expressed in multiple cell types including Cardiomyocytes, Vascular Endothelial cells, and Vascular Smooth Muscle Cells. Three major MAPKs include ERKs (Extracellular signal-Regulated Kinases), JNKs (c-Jun NH(2)-terminal protein Kinases), and p38 Kinases. Members of the JNK/SAPK (Stress-Activated Protein Kinase) family of MAPKs are strongly stimulated by numerous Environmental Stresses, but also more modestly stimulated by Mitogens, Inflammatory Cytokines, Oncogenes, and inducers of Cell differentiation and morphogenesis. Ten mammalian JNK isoforms have been identified and are encoded by three distinct genes, JNK1, JNK2, and JNK3, the transcripts of which are alternatively spliced to yield four JNK1 isoforms, four JNK2 isoforms, and two JNK3 isoforms. JNK1 and JNK2 are the products of alternative splicing of a single gene and are expressed in many tissues, but JNK3 is specifically expressed in brain. Members of the JNK family play crucial roles in regulating responses to various Stresses, and in Neural Development, Inflammation, and Apoptosis. JNK activation is much more complex than that of ERK1/ERK2 owing to inputs by a greater number of MAPKKKs (Mitogen-Activated Protein Kinase Kinase Kinases) (at least 13, including MEKK1 (MAP/ERK Kinase-Kinase-1)-MEKK4 (MAP/ERK Kinase-Kinase-4), ASK (Apoptosis Signal-regulating Kinase) and MLKs (Mixed-Lineage Kinases), which are activated by upstream Rho-family GTPases). These activate JNK MAPKKs MEK4 (MAPK/ERK Kinase-4) and MEK7 (MAPK/ERK Kinase-7), which further activate JNKs. The JNK MAPK modules are regulated by a number of different scaffold proteins, including JIP1 (JNK Interacting Protein-1), JIP2 (JNK Interacting Protein-2), JIP3 (JNK Interacting Protein-3), JIP4 (JNK Interacting Protein-4), Beta-Arrestin-2, Filamin and CrkII. The scaffold proteins presumably target the MAPK modules to different sites in the cell and play roles in kinase activation and/or substrate selection (Ref.1 & 2).
Stress or Genotoxic agents are the most powerful inducers of JNK. Different forms of stress have been shown to mediate JNK activation via various cellular pathways. JNK activation in response to UV irradiation is mediated by upstream signaling components, including Rac (Ras-Related C3 Botulinum Toxin Substrate), CDC42 (Cell Division Cycle-42), PAK (p21/CDC42/Rac1-Activated Kinase), ASK1 (Apoptosis Signal-regulating Kinase-1), MLK, MEKK1, SEK1 (SAPK/ERK Kinase-1)/MKK4, MKK7 and p21Ras, in concert with nuclear DNA lesions. Besides Stress, JNKs can also be activated via GPCRs (G-Protein Coupled Receptors), RTKs (Receptor Tyrosine Kinases) and Cytokine Receptors. How GPCRs activate the JNKs is still an unanswered question. Free Beta-Gamma dimers and GN-Alpha12 and GN-Alpha13 proteins are able to activate JNK in a Rac1-CDC42 or p115RhoGEF and RhoA-dependent manner. However, the nature of the GEFs (Guanine nucleotide Exchange Factors) that connect Beta-Gamma and GN-Alpha12/ GN-Alpha13 to Rac1 and CDC2 is still unclear. Interestingly, GN-Alpha12 can also activate JNK by activating the MEKK (MEK kinase). The activation of JNK by Cytokine receptors appears to be mediated by the TRAF (TNF Receptor-Associated Factor) group of Adaptor proteins. Activation of the TNF receptor leads to recruitment of TRAF2 (TNF Receptor-Associated Factor-2), which is required for JNK activation. These adaptor proteins (TRADD (Tumor Necrosis Factor Receptor-1-Associated Death Domain Protein), RIP (Receptor-Interacting Protein) and Daxx) have been reported to bind MEKK1 and ASK1. TRAF2 activates MAPK4Ks like GCK (Germinal Center Kinase), GCKR (GCK-Related Kinase), GLK (GCK-Like Kinase) and HGK (HPK/GCK-like Kinase), which further activates JNKs via MEKK1 and MKK4/7 respectively. ASK1 also interacts with TRAF2 and activates JNK via MKK4/7 (Ref. 3, 4 & 5).
Growth Factors also activate JNKs. Although the Signaling cascade from Growth Factor Receptors to ERKs is relatively well understood, the pathway leading to JNK activation is more obscure. Activation of JNK by EGF (Epidermal Growth Factor) or NGF (Nerve Growth Factor) is dependent on H-Ras activation. Growth Factors and Growth Factor Receptors stimulate Ras by recruiting SOS (Son of Sevenless), GRB2 (Growth Factor Receptor-Bound Protein-2) and SHC to the membrane. PI3K (Phosphatidylinositde-3-Kinase) also activate Ras. Ras activates two protein kinases, Raf1 and MEKK (MEK (MAPK, or ERK, kinase) Kinase). Raf1 contributes directly to ERK activation but not to JNK activation, whereas MEKK participated in JNK activation but caused ERK activation only after overexpression. Recently, Raf1 is found to interact with the proapoptotic, stress-activated protein kinase ASK1 in vitro and in vivo. This interaction allows Raf1 to act independently of the MEK–ERK pathway to activate JNK pathway (Ref.6 & 7). The Rho family GTPases, CDC42 (Cell Division Cycle-42) and Rac also initiate a cascade leading to JNK/SAPK, presumably by binding and activating the protein kinase PAK (p21-Activated Kinases), a kinase that phosphorylates and promotes activation of MEKK1. Rac/CDC42 are also involved in JNK activation via a pathway consisting of a sequential cascade MLKs and MKK4/7 (MAP Kinase Kinase-4/7. MLK2 (Mixed-Lineage Kinase-2) and MLK3 (Mixed-Lineage Kinase-3) interact with the activated (GTP-bound) forms of Rac and CDC42, with a slight preference for Rac. Besides MLKs, MEKK1/4 and Posh (Plenty of SH3) are also activated by Rac/CDC42 to activate MKK4/7 and thus JNKs. Adaptor proteins such as Crk (v-Crk Avian Sarcoma Virus Ct10 Oncogene Homolog) and CrkL (v-Crk Avian Sarcoma Virus Ct10 Oncogene Homolog-Like) also leads to activation of JNKs in response to RTK. HPK1 (Hematopoietic Progenitor Kinase-1) associates with Crk and CrkL through binding to the SH3 (Src-Homology Domain-3) of these proteins. Furthermore, association of HPK1 with these proteins increases HPK1's kinase activity. HPK1 then act as upstream of MEKK1 and TAK1 (Transforming Growth Factor-Beta-activated Kinase-1) in the JNK kinase cascade. JNKs are negatively regulated by MKP (MAP Kinase Phosphatase) (Ref.2, 8 & 9).
The activated JNK/SAPKs translocate to the nucleus where they phosphorylate transcription factors such as c-Jun, Elk1, DPC4 (Deleted In Pancreatic Carcinoma 4)/ SMAD4 (Sma and MAD (Mothers Against Decapentaplegic) Related Protein-4), p53, ATF2 (Activating Transcription Factor-2), NFAT4 (Nuclear Factor of Activated T-Cell-4) and NFAT1 (Nuclear Factor of Activated T-Cell-1). JNK1 directly phosphorylates Bcl2 (B-Cell CLL/Lymphoma-2) in vitro, co-localizes and collaborates with Bcl2 to mediate prolonged cell survival. JNK cascade also activates TCF (Ternary Complex Factor) protein. JNK also phosphorylate HSF1 (Heat Shock Factor-1) and JNK-mediated phosphorylation of HSF1 selectively stabilize the HSF1 protein and confers protection to cells under conditions of severe stress. DCX is also a substrate of JNK and interacts with both JNK and JIP. MAPs (Microtubule-Associated Proteins), both MAP1B and MAP2B are also found to be the substrates of JNK. Ser-727 phosphorylation of STAT3 (Signal Transducer and Activator of Transcription-3) can also be induced by JNK. JNK also regulates Insulin signaling by negatively regulating IRS1 (Insulin Receptor Substrate-1). JNK is generally thought to be involved in inflammation, proliferation and Apoptosis. Accordingly, its substrates are transcription factors and Anti-apoptotic proteins. However, JNK also phosphorylates Serine 178 on Paxillin and regulate cell migration. Despite extensive progress in the understanding of the JNK MAP kinase pathway, the mechanisms by which the pathway contributes to the many cellular programs where JNKs are activated are poorly defined. The JIP1 proteins have been proposed to act as molecular scaffolds that organize the JNK signal transduction pathway in response to specific stimuli. The JNK stress pathway is thought to be important in many pathological conditions including the progression of some neurodegenerative diseases such as Huntington’s and also in cancer. This pathway therefore offers potential targets for therapeutic intervention. The identification of critical components of this signaling pathway, such as JIP1, offers new routes to understand how this pathway is regulated and potential ways of manipulating it to combat disease (Ref.10, 11 & 12).
References
- 1
- Himes SR, Sester DP, Ravasi T, Cronau SL, Sasmono T, Hume DA. The JNK are important for development and survival of macrophages.
- 2
- Moulin N, Widmann C. Islet-brain (IB)/JNK-interacting proteins (JIPs): future targets for the treatment of neurodegenerative diseases?
- 3
- Zhou JY, Liu Y, Wu GS. The role of mitogen-activated protein kinase phosphatase-1 in oxidative damage-induced cell death.
- 4
- Yang L, Mao L, Chen H, Catavsan M, Kozinn J, Arora A, Liu X, Wang JQ. A signaling mechanism from G alpha q-protein-coupled metabotropic glutamate receptors to gene expression: role of the c-Jun N-terminal kinase pathway.
- 5
- Yang Q, Kim YS, Lin Y, Lewis J, Neckers L, Liu ZG. Tumour necrosis factor receptor 1 mediates endoplasmic reticulum stress-induced activation of the MAP kinase JNK.
- 6
- Kraus S, Benard O, Naor Z, Seger R. c-Src is activated by the epidermal growth factor receptor in a pathway that mediates JNK and ERK activation by gonadotropin-releasing hormone in COS7 cells.
- 7
- Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response.
- 8
- Yamauchi J, Miyamoto Y, Kokubu H, Nishii H, Okamoto M, Sugawara Y, Hirasawa A, Tsujimoto G, Itoh H. Endothelin suppresses cell migration via the JNK signaling pathway in a manner dependent upon Src kinase, Rac1, and Cdc42.
- 9
- Zhou JY, Liu Y, Wu GS. The role of mitogen-activated protein kinase phosphatase-1 in oxidative damage-induced cell death.
- 10
- Baan B, van Dam H, van der Zon GC, Maassen JA, Ouwens DM. The role of JNK, p38 and ERK MAP-kinases in insulin-induced Thr69 and Thr71-phosphorylation of transcription factor ATF2.
 关于我们
关于我们