Interferon_Pathway
发布时间:2019-12-10 19:07 来源:SABiosciences
- 通路
- 概述
Review
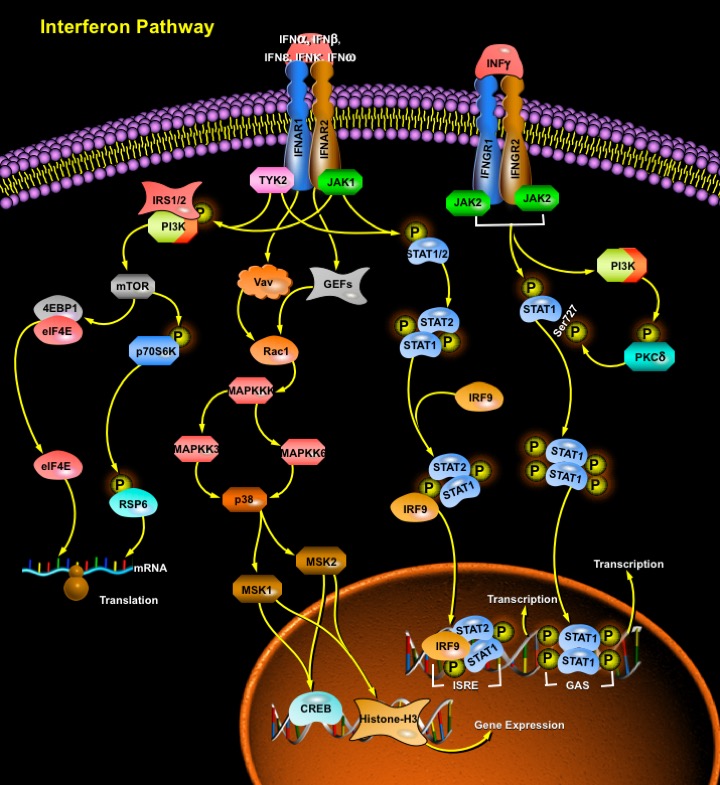
To thwart viral infection, our cells have developed a formidable and integrated defense network that comprise of innate and adaptive immune responses. In an attempt to prevent viral replication, viral dissemination or persistent viral infection of the cell, many of these protective measures actually involve the induction of programmed cell death, or apoptosis. Once the virus has invaded the cell, a host defense-mediated response is triggered which involves the induction of a family of pleiotropic cytokines known as the IFNs (Interferons) (Ref.1). These IFNs constitute a heterogeneous group of proteins and are best known for their ability to induce cellular resistance to virus infection. However, IFNs also affect many other cellular functions, such as cell growth. IFNs are classified as either Type-I or Type-II. There are many Type-I IFNs, all of which have considerable structural homology. These include IFN-Alpha (which can be further subdivided into 13 different subtypes, IFN-Alpha1, -Alpha2, -Alpha4, -Alpha5, -Alpha6, -Alpha7, -Alpha8, -Alpha10, -Alpha13, -Alpha14, -Alpha16, -Alpha17 and -Alpha21), IFN-Beta, IFN-Delta, IFN-Epsilon, IFN-Kappa, IFN-Tau and IFN-Omega (Ref.2). IFN-Alpha, IFN-Beta, IFN-Epsilon, IFN-Kappa and IFN-Omega exist in humans, whereas IFN-Delta and IFN-Tau have been described only for pigs and cattle, respectively, and do not have human homologues (Ref.3, 19 & 20). All Type-I IFNs bind a common cell-surface receptor, which is known as the Type-I IFN receptor. By contrast, there is only one Type-II IFN, IFN-Gamma (Ref.2). IFN-Gamma binds a different cell-surface receptor, which is known as the Type-II IFN receptor. IFN-Gamma is a markedly different cytokine than the Type-I IFNs, but it was originally classified in the IFN family because of its ability to ‘interfere’ with viral infections, which is consistent with the original definition of IFNs. Recently, a new class of IFNs or IFN-like molecules has emerged, the IFN-Lambda molecules: IFN-Lambda1, -Lambda2 and –Lambda3, which are also known as IL-29 (Interleukin-29), IL-28A and IL-28B, respectively (Ref.4, 13, 14 & 15). They also have antiviral properties, but they are distinct from the Type-I and Type-II IFNs and bind a different cell-surface receptor, which is composed of two chains, IFNLR1 (also known as IL-28 Receptor-Alpha, IL-28RAlpha) and IL-10RBeta. IFN-Lambda molecules might ultimately be classified and accepted as Type-III IFNs. Viral infection elicits the production of Type-I IFNs, which upon interaction with their receptor induce a set of IFN-stimulated genes that inhibit viral replication and increase the lytic potential of NK (Natural Killer) cells. In addition to this role in innate immunity, Type-I IFNs modulate the adaptive immune response by increasing MHC-I (Major Histocompatibility Complex Class-I) expression to promote antigen presentation, also promoting T-Cell survival and stimulating dendritic cell maturation (Ref.1).
Both the Type-I IFN receptor and the Type-II IFN receptor have multichain structures, which are composed of at least two distinct subunits: IFNAR1 and IFNAR2 for the Type-I IFN receptor, and IFNGR1 and IFNGR2 for the Type-II IFN receptor. Each of these receptor subunits interacts with a member of the JAK (Janus Activated Kinase) family. In the case of the Type-I IFN receptor, the IFNAR1 subunit is constitutively associated with TYK2 (Tyrosine Kinase-2), whereas IFNAR2 is associated with JAK1 (Ref.5, 6 & 7). In the case of the Type-II IFN receptor, the IFNGR1 subunit associates with JAK1, whereas IFNGR2 is constitutively associated with JAK2 (Ref.6). The initial step in both Type-I- and Type-II-IFN-mediated signaling is the activation of these receptor-associated JAKs, which occurs in response to a ligand-dependent rearrangement and dimerization of the receptor subunits, followed by autophosphorylation and activation of the associated JAKs. The binding of IFN-Alpha or other Type-I IFNs to the Type-I IFN receptor results in the rapid autophosphorylation and activation of the receptor associated JAKs TYK2 and JAK1 (Ref.8 & 4), which in turn regulate the phosphorylation and activation of STATs. The STATs that are activated in response to Type-I IFNs include STAT1, STAT2, STAT3 and STAT5. The activation of such STATs is a common response to different Type-I IFNs, consistent with all of these IFNs binding the same receptor and thereby activating a common pathway that involves the same JAKs, TYK2 and JAK1. STAT4 and STAT6 can also be activated by IFN-Alpha, but such activation seems to be restricted to certain cell types, such as endothelial cells or cells of lymphoid origin (Ref.9 & 10). After phosphorylation by JAKs, the activated STATs form homodimers or heterodimers that translocate to the nucleus and initiate transcription by binding specific sites in the promoters of ISGs (IFN-Stimulated Genes) (Ref.6). An important transcriptional complex that is induced by Type-I IFNs is the ISGF3 (ISG Factor-3) Complex. The mature ISGF3 Complex is composed of the phosphorylated (activated) forms of STAT1 and STAT2, together with IRF9, which does not undergo tyrosine phosphorylation (Ref.11, 13, 14 & 15). This complex is the only complex that binds specific elements known as ISREs (IFN-Stimulated Response Elements) that are present in the promoters of certain ISGs, thereby initiating their transcription. Other STAT complexes that are induced by Type-I IFNs include STAT1–STAT1, STAT3–STAT3, STAT4–STAT4, STAT5–STAT5 and STAT6–STAT6 homodimers, as well as STAT1–STAT2, STAT1–STAT3, STAT1–STAT4, STAT1–STAT5, STAT2–STAT3 and STAT5–STAT6 heterodimers (Ref.11 & 12). Such IFN-induced complexes bind another type of element — known as GAS (IFN-Gamma-Activated Site) element—that is present in the promoter of ISGs (Ref.11). Of the hundreds of known ISGs, some have only ISREs or only GAS elements in their promoters, whereas others have both elements; therefore, combinations of different STAT-containing complexes might be required for the optimal transcriptional activation of a particular gene. The transcription of Type-II IFN (IFN-Gamma)-dependent genes is regulated by GAS elements, and STAT1 is the most important IFN-Gamma-activated transcription factor for the regulation of these transcriptional responses. After engagement of the Type-II IFN receptor by IFN-Gamma, JAK1 and JAK2 are activated and regulate downstream phosphorylation of STAT1 on the tyrosine residue at position Tyr701 (Ref.11). The tyrosine phosphorylated form of STAT1 forms homodimers that translocate to the nucleus and bind GAS elements, which are present in the promoters of IFN-Gamma-regulated genes. The IFN-Gamma-activated JAKs also regulate, through as yet- unknown intermediates, activation of the catalytic subunit (p110) of PI3K. The activation of PI3K ultimately results in downstream activation of PKC-Delta (Protein Kinase-C-Delta), which in turn regulates phosphorylation of STAT1 on Ser727. The phosphorylation of Serine is not essential for the translocation of STAT1 to the nucleus or for the binding of STAT1 to DNA, but it is required for full transcriptional activation. Phosphorylated STAT1 homodimerizes to form the GAF-AAF complex, which translocates to the nucleus and binds to the GAS element present in most IFN-Gamma inducible genes. In contrast to Type-I IFNs, IFN-Gamma does not induce the formation of ISGF3 complexes and thereby cannot induce the transcription of genes that have only ISREs in their promoter. Like IFN-Gamma, IFN-Alpha and IFN-Beta signaling can also lead to the formation of the GAF-AAF complex and it’s binding to the GAS regulatory element. The three newly identified IFN-Lambda proteins (also termed IL-28A, IL-28B and IL-29) bind to a heterodimeric Class-II cytokine receptor subunit IFN-LamdaR1 (also termed CRF2-12) and a second subunit IL-10R2 (CRF2-4), which serves as the second chain of the IL-10R. IFN-Lambda cross-linking leads to both STAT1 and STAT2 activation resulting in the formation of ISGF3 and GAF-AAF complexes. These newly described cytokines are functionally similar to the Type-I IFNs because their synthesis is induced by virus infection or double-stranded RNA. They render cells resistant to virus infection and activate the same intracellular signaling pathways as Type-I IFNs. The IFN-Lambda family represents an interesting evolutionary link between the Type-I IFNs and the IL-10 family but they are structurally more closely related to the Type-I IFNs. IFN-Lambda also activates STAT3 and STAT5, which is more characteristically associated with signaling by IL-10 and IL-10 related cytokines (Ref.3).
Another signaling cascade regulated by IFNs is the MAPK (Mitogen Activated Protein Kinase) pathway (Ref.16). IFN-activated JAKs regulate the phosphorylation of Vav or other GEFs (Guanine-Nucleotide-Exchange Factors), resulting in the downstream activation of Rac1 that can regulate the signaling pathway of the p38MAPK. A MAPKKK (MAPK Kinase Kinase) is subsequently activated and regulates downstream activation of the MAPK kinases MAPKK3 and MAPKK6, which directly phosphorylate p38, resulting in its activation. Activated p38 subsequently regulates activation of multiple downstream effectors, including MAPKAPK2 (MAPK-Activated Protein Kinase-2), MAPKAPK3, MSK1/2 (Mitogen-and Stress-Activated Kinase) and MNK1 (MAPK-Interacting Protein Kinase-1) (Ref.17). The specific transcription factors that are regulated by p38s include CREB (cAMP Responsive Element Binding protein) and Histone-H3. Activated TYK2 and JAK1 regulate tyrosine phosphorylation of IRS1 and IRS2 (Insulin Receptor Substrate), which provide docking sites for the SH2 (SRC Homology-2) domains of the regulatory subunit (p85) of PI3K (Phosphatidylinositol 3-Kinase). PI3K is subsequently activated and regulates downstream activation of mTOR (Mammalian Target of Rapamycin) which mediates the initiation of mRNA translation. In turn, mTOR regulates phosphorylation (activation) of p70S6K (p70S6 Kinase), which then phosphorylates RPS6 (Ribosomal Protein S6), resulting in the initiation of mRNA translation. mTOR also regulates phosphorylation of the translational repressor 4EBP1 (Eukaryotic Translation-Initiation Factor 4E (EIF4E)-Binding Protein-1). Such phosphorylation results in its de-activation and subsequent dissociation from EIF4E, allowing the initiation of cap-dependent mRNA translation.
The Type-I IFNs are efficacious in the treatment of malignancies, viral infections and autoimmune diseases. IFN-Alpha plays a role in the management of different neoplasias, particularly those of hematological origin, i.e. of a myeloid and lymphoid etiology (Ref.17). IFN-Beta is the only interferon that has been shown to have beneficial effects in the treatment of MS (Multiple Sclerosis). IFN-Gamma is an absolute requirement for resistance against acute acquired infection with Toxoplasma gondii and development of TE (Toxoplasmic Encephalitis) during the late stage of infection (Ref.18). Thus, this unique repertoire of biological activities of IFNs has generated considerable interest in its potential for the treatment of viral infections, cancers and immunological disorders. Indeed, administration of IFNs is now the treatment of choice for some cancers, particularly Leukemias and Kaposi Sarcoma, Rheumatoid Arthritis and viral infections such as Chronic Hepatitis-B and C. However, many other types of cancer and viral infections are only partially responsive to IFN therapy. The molecular mechanisms, which determine the antiviral and antiproliferative sensitivity or resistance to IFNs, have still to be fully elucidated.
References
- 1
- Barber GN. Host defense, viruses and apoptosis.
- 2
- Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors.
- 3
- Vilcek J. Novel interferons.
- 4
- Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex.
- 5
- Nguyen KB, Cousens LP, Doughty LA, Pien GC, Durbin JE, Biron CA. Interferon alpha/beta-mediated inhibition and promotion of interferon gamma: STAT1 resolves a paradox.
- 6
- Chen J, Baig E, Fish EN. Diversity and relatedness among the type I interferons.
- 7
- Katsoulidis E, Li Y, Mears H, Platanias LC. The p38 mitogen-activated protein kinase pathway in interferon signal transduction.
- 8
- Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, Ostrander C, Dong D, Shin J, Presnell S, Fox B, Haldeman B, Cooper E, Taft D, Gilbert T, Grant FJ, Tackett M, Krivan W, McKnight G
- 9
- Farrar JD, Smith JD, Murphy TL, Murphy KM. Recruitment of Stat4 to the human interferon-alpha/beta receptor requires activated Stat2.
- 10
- Torpey N, Maher SE, Bothwell AL, Pober JS. Interferon alpha but not interleukin 12 activates STAT4 signaling in human vascular endothelial cells.
 关于我们
关于我们