iNOS_Signaling
发布时间:2019-12-10 16:21 来源:SABiosciences
- 通路
- 概述
Review
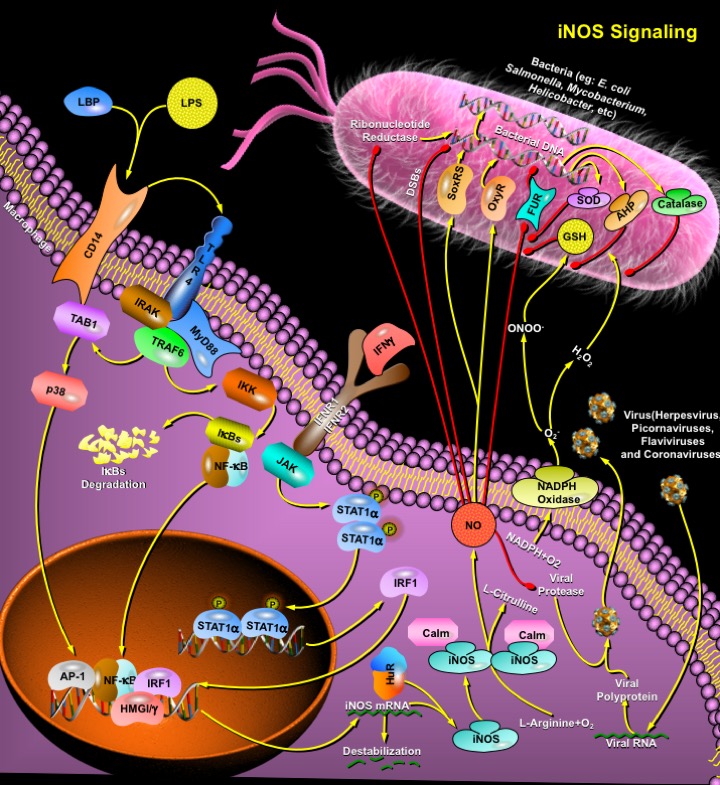
Microorganisms have developed several mechanisms to survive in their hosts' environments. These include competition with their hosts for metal acquisition and resistance to host defenses such as NO (Nitric Oxide), a cytotoxic weapon generated by macrophages. In eukaryotic cells, NO is metabolically produced by NOS (NO Synthase) from L-Arginine, O2 (Molecular Oxygen), and NADPH (Nicotinamide Adenine Dinucleotide, Reduced). In macrophages, an inducible NO synthase (iNOS or NOS2) is produced after activation by endotoxins or cytokines and generates copious amounts of NO presumably to help kill or inhibit the growth of invading microorganisms or neoplastic tissue (Ref.1 & 2). Although iNOS was originally identified and characterized in macrophages, it is present in numerous cell types including endothelial cells, fibroblasts, vascular smooth muscle cells and cardiac myocytes. Catalytic activity of iNOS is regulated by the availability of the substrate, Calm (Calmodulin), L-Arginine, and of the cofactors, NADPH and tetrahydrobiopterin. NOS2 utilizes oxygen and electrons from NADPH to oxidize the substrate L-Arginine into the intermediate OH-L-Arginine, which is then oxidized into NO and L-Citrulline. NO and superoxide (O2-) are radical effectors of the innate immune system that can directly inhibit pathogen replication.
Although NOS2 activity is independent of calcium concentrations, a variety of extracellular stimuli can activate distinct signaling pathways that converge to initiate expression of iNOS. Cell wall components of bacteria and fungi trigger the innate immune signaling cascade, leading to expression of iNOS. LPS (Lipopolysaccharide), a component of the wall of Gram-negative bacteria, binds to LBP (LPS-Binding Protein), which delivers LPS to CD14, a high-affinity LPS receptor. TLR4 (Toll-Like Receptor-4) in conjunction with the small extracellular protein MD2 interacts with the CD14-LPS complex, and then activates an intracellular signaling cascade via adaptors that include IRAK (Interleukin-1 Receptor-Associated Kinase) and MyD88 (Myeloid Differentiation Primary Response Gene-88). These adaptors in turn activate downstream molecules including TRAF6 (TNF Receptor-Associated Factor-6), TAB1 (TAK1-Binding Protein-1) and p38. LPS activation of TLR4 leads to phosphorylation of IKK (Inhibitor of KappaB Kinase), which phosphorylates the I-kappaB and releases the transcription factor NF-kappaB (Nuclear Factor-KappaB). NF-kappaB translocates from the cytoplasm to the nucleus, where it interacts with kappaB elements in the NOS2 5' flanking region, triggering NOS2 transcription (Ref.2, 3 & 8). Cytokines released from infected host cells also activate NO production, including TNF-Alpha (Tumor Necrosis Factor-Alpha) and IL-1Beta (Interleukin-1Beta). IFN-Gamma (Interferon-Gamma) interacts with the IFNR1 (Interferon Receptor-1) and IFNR2 complex, which activates kinases of the JAK (Janus Kinase) family and STAT (Signal Transducers and Activators of Transcription) pathways leading to synthesis of the transcription factor IRF1 (Interferon Response Factor-1) and stimulation of NOS2 mRNA transcription. IFN-Gamma also provides a synergistic boost to LPS induction of NOS2 transcription because IRF1 interacts with NF-kappaB, altering the conformation of the NOS2 promoter. Nuclear proteins that interact with members of the NF-kappaB family include the nonhistone chromosomal proteins, HMGI/Y (High Mobility Group Family). They enhance the binding of transcription factors, such as NF-kappaB and AP-1 (Activating Protein-1), to their binding sites by DNA-protein and protein-protein interactions (Ref.3 & 5). Other transcription factors, including STAT1Alpha and HIF1 (Hypoxia Inducible Factor-1) also regulate NOS2 expression.
NO is an anti-bacterial effector and can inhibit bacterial DNA synthesis by inhibiting bacterial Ribonucleotide Reductase1/2 and causing DSBs (Double-Stranded Breaks) in bacterial DNA. It can also increase the susceptibility of bacteria to oxidative DNA damage by blocking respiration. NO combines with O2- to form ONOO- (peroxynitrite anion) and oxidize bacterial lipids to produce nitrotyrosine, but the biological significance of these modifications is still unclear. Some bacteria contain low concentrations of GSH (Glutathione), and are susceptible to NO. The bacterial protein SoxRS (Superoxide Regulon) serves as a sensor for NO, and can activate transcription of a set of bacterial genes whose products defend the pathogen from oxidant damage by bacterial SOD (Superoxide Dismutase). OxyR (Peroxide Regulon), a transcription factor involved in stimulation of peroxide detoxification genes, is directly modified by H2O2 (Hydrogen Peroxide) or NO via S-nitrosylation and assists in protecting the bacterium from the NO donor S-nitrosocysteine. It also directs the transcription of bacterial genes such as AHP (Alkyl Hydroperoxide Reductase), which confers resistance, to peroxynitrite, and Catalase, which deactivates H2O2 (Ref.4 & 9). The bacterial protein FUR (Ferric Uptake Regulatory protein) also serves as an NO sensor. NO inactivates FUR by interacting with its iron cofactor, permitting expression of genes protective against oxidative stress. One bacterial gene regulated by FUR encodes a flavohemoglobin that can detoxify NO, protecting pathogens from NO. Thus multiple signaling pathways defend bacteria against NO. NO is also an anti-viral effector of the innate immune system. It can inhibit replication of Herpes viruses, Picornaviruses, Flaviviruses and Corona viruses by targeting viral proteases. Many RNA viruses depend on viral proteases to cleave large viral polyproteins into smaller viral polypeptides. In Toxoplasma gondii infection, the induction of iNOS serves as a nonspecific immune response that prevents parasite invasion.
While iNOS induction can protect brain from certain infectious diseases, excessive levels of NO can also be toxic to neurons. Increased NO production via induction of iNOS has been suggested as a major mechanism by which cytokines mediate cardiac contractile dysfunction and development of cardiovascular disease. Over-expression of iNOS, a common phenomenon during chronic inflammatory conditions, generates sustainable amounts of NO, and its reactive intermediates are mutagenic, causing DNA damage or impairment of DNA repair. Recent studies also implicate NO as having a key signaling molecule that regulates processes of tumorigenesis. Increased expression of iNOS has been involved in tumors of the colon, lung, oropharynx, reproductive organs, breast, and CNS (Central Nervous System) besides its occurrence in chronic inflammatory diseases. Development of selective inhibitors of iNOS and NO-releasing agents may lead to important strategies for chemoprevention of cancer (Ref.6, 7 & 9).
References
- 1
- Petruson K, Stalfors J, Jacobsson KE, Ny L, Petruson B. Nitric oxide production in the sphenoidal sinus by the inducible and constitutive isozymes of nitric oxide synthase.
- 2
- Kadowaki S, Chikumi H, Yamamoto H, Yoneda K, Yamasaki A, Sato K, Shimizu E. Down-regulation of inducible nitric oxide synthase by lysophosphatidic acid in human respiratory epithelial cells.
- 3
- Davis RL, Sanchez AC, Lindley DJ, Williams SC, Syapin PJ. Effects of mechanistically distinct NF-kappaB inhibitors on glial inducible nitric-oxide synthase expression.
- 4
- Saldeen J, Welsh N. p38 MAPK inhibits JNK2 and mediates cytokine-activated iNOS induction and apoptosis independently of NF-KB translocation in insulin-producing cells.
- 5
- Jang BC, Paik JH, Kim SP, Bae JH, Mun KC, Song DK, Cho CH, Shin DH, Kwon TK, Park JW, Park JG, Baek WK, Suh MH, Lee SH, Baek SH, Lee IS, Suh SI. Catalase induces the expression of inducible nitric oxide synthase through activation of NF-kappaB and PI
- 6
- Gavrilescu LC, Butcher BA, Del Rio L, Taylor GA, Denkers EY. STAT1 is essential for antimicrobial effector function but dispensable for gamma interferon production during Toxoplasma gondii infection.
- 7
- Lala PK, Chakraborty C. Role of nitric oxide in carcinogenesis and tumour progression.
- 8
- Mizel SB, Honko AN, Moors MA, Smith PS, West AP. Induction of macrophage nitric oxide production by Gram-negative flagellin involves signaling via heteromeric Toll-like receptor 5/Toll-like receptor 4 complexes.
- 9
- Bafica A, Scanga CA, Serhan C, Machado F, White S, Sher A, Aliberti J. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production.
 关于我们
关于我们