ILK_Signaling
发布时间:2019-12-10 16:17 来源:SABiosciences
- 通路
- 概述
Review
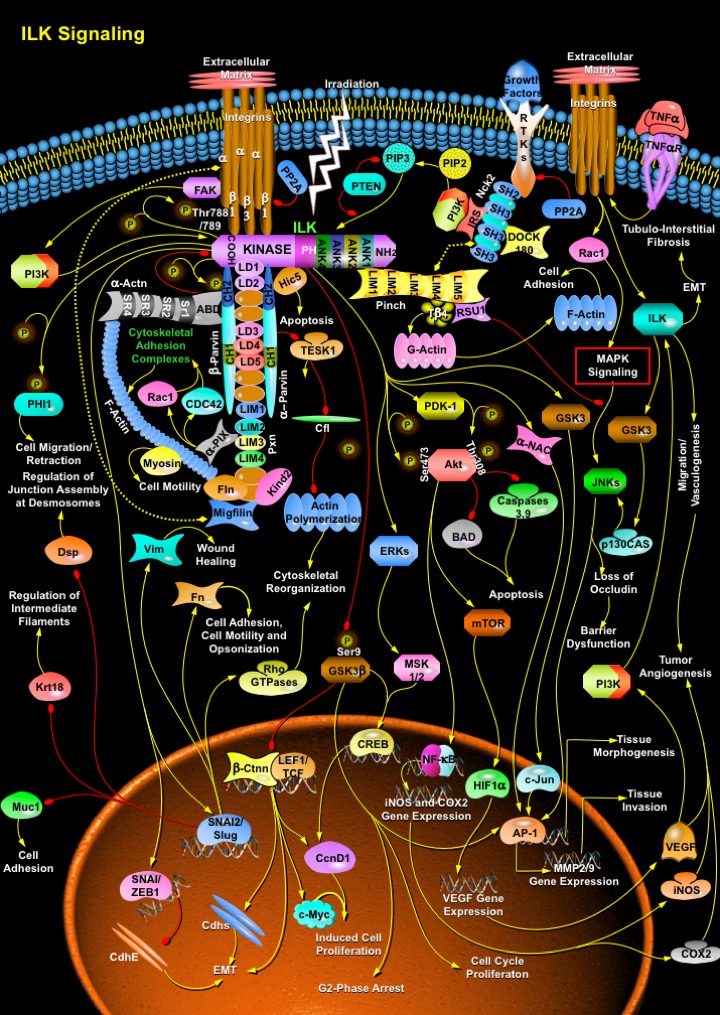
The ECM (Extracellular Matrix) provides the structural framework for the formation of tissues and organs. The ECM binds to substrate adhesion molecules on the surface of cells and influences various intracellular signaling pathways that regulate survival, proliferation, polarity and differentiation. The important families of adhesion molecules that bind to the ECM are the Integrins. Integrins consist of Alpha and Beta-subunits and are composed of large extracellular domains and relatively small cytoplasmic domains (Ref.1 & 2). Ligand binding activates signaling cascades that lead to the assembly of a multiprotein complex at the site of cell adhesion to the ECM. These events have two important impacts on the cell: they forge a connection between the ECM and the Actin cytoskeleton, and they alter the fluxes of many intracellular signaling pathways. Three proteins which act as important regulators of Integrin mediated signaling are the ILK (Integrin Linked Kinase) and the adaptor proteins PINCH (Particularly Interesting Cys-His-rich Protein) and Parv (Parvin), among which ILK is the major regulator of Integrin mediated signaling. These molecules form a heterotrimeric complex, which is referred to as the IPP complex and is named after its components in order of their discovery. The IPP complex functions both as an adaptor between Integrins and the Actin cytoskeleton and as a hub that regulates several signaling pathways. The main function of ILK is to connect Integrins to the cytoskeleton. ILK recruits other adaptor molecules into a large complex to regulate Actin dynamics and Integrin function (Ref.2).
Several adaptor proteins with Actin binding properties interact with the C-terminus (COOH-terminus) of ILK. ILK consists of three domains, ANK (N-terminal Ankyrin) repeats, a PH (Plekstrin Homology) domain and a C-terminal Kinase domain. The ANK repeats of ILK are four in number. ANK1 binds to the LIM1 domain of PINCH isoforms as well as to the ILKAP (ILK Associated Phosphatase). ILKAP, a Serine/Threonine phosphatase, negatively regulates ILK activity and ILK signaling. The PH domain of ILK probably binds to PIP3 (Phosphatidylinositol-3,4,5-trisphosphate). The tumor suppressor PTEN (Phosphatase and Tensin Homolog) dephosphorylates PIP3 to PIP2 (Phosphatidylinositol-4,5-diphosphate), resulting in the inhibition of ILK activity (Ref.3). The kinase domain of ILK binds to the cytoplasmic tails of Beta-Integrins, the kinase substrate Akt (v-Akt Murine Thymoma Viral Oncogene Homolog), the PDK-1 (Phosphoinositide-Dependent Kinase-1), Parvins, Kind2 (Kindlin-2)/MIG2 (Mitogen-Inducible Gene-2 Protein) and Pxn (Paxillin). Pxn contains both LIM and LD (Leucine Rich Domains). The LD domains of Pxn interact with the CH (Calponin Homology) domains of Alpha-Parvin and Beta-Parvin, which then mediate direct interaction with Actin. Alpha- and Beta-Parvins bind to F-Actin directly, as well as indirectly through binding to Pxn or Hic5 (Alpha-Parvin only) or Alpha-Actinin (Beta-Parvin only). Alpha-Actinin binds to Beta-Parvin and F-Actin through its ABD (Actin Binding Domain) and SR (Spectrin Repeats), respectively. Alpha-Parvin also binds to TESK1 (Ser/Thr Kinase Testicular Protein Kinase-1), whereas Beta-Parvin binds to the guanine nucleotide-exchange factor PIX-Alpha (PAK-Interacting Exchange Factor-Alpha), which influences Actin remodeling through the GTPases, Rac1 (Ras-Related C3 Botulinum Toxin Substrate-1) and CDC42 (Cell Division Cycle-42) by activating the cytoskeletal adhesion complexes. Interactions with Integrins and the cytoskeleton (Actin and Myosin) also occur through a MIG2/Kind2-Migfilin-Fln (Filamin) complex and promote cell motility (Ref.2). TESK1 mainly phosphorylate Cfl (Cofilin) and induce Actin cytoskeletal reorganization, whereas, Hic5 regulates Apoptosis. Phosphorylation of the N-terminus of Alpha-Parvin increases binding to ILK and inhibits binding to TESK1. Since Alpha-Parvin inhibits TESK1 activity, release of TESK1 upon phosphorylation of Alpha-Parvin results in inhibition of Cfl by phosphorylation and consequently, reduction of the turnover rates of Actin filaments. Alpha-Parvin may bind to F-Actin in vitro but the functional significance of this interaction remains to be established. The PINCH-ILK-Alpha-Parvin complex plays a critical role in cell survival by promoting membrane recruitment of Akt and its activation by phosphorylation. Alpha-Parvin and Beta-Parvin compete for binding to ILK and have opposite effects on ILK kinase activity. While Alpha-Parvin binding increases ILK activity, Beta-Parvin binding represses it. Also, Beta-Parvin promotes adhesion and spreading through binding of Alpha-Actinin, Alpha-PIX and activation of Rac1, while Alpha-Parvin represses Rac1 (Ras-Related C3 Botulinum Toxin Substrate-1) (Ref.4).
ILK binds PINCH through its N-terminal domain and binds Alpha-Parvin or Beta-Parvin through its C-terminal domain, resulting in formation of PINCH-ILK-Parvin ternary complexes. Formation of these complexes occurs before localization to Integrin rich adhesion sites and is required for preventing degradation of ILK, PINCH and Parvins. Recruitment of these complexes to Integrin rich adhesion sites is mediated by interactions of the complexes with additional proteins, such as Beta1-Integrin, MIG2/Kind2 and Pxn. Finally, ILK connects via PINCH to NCK2, known to bind to DOCK180 (Dedicator of Cytokinesis-180), another guanine nucleotide exchange factor mediating Actin cytoskeleton dynamics via Rac1 (Ref.5). PINCH isoforms, which contain five LIM domains, bind to RTKs (Receptor Tyrosine Kinases) through the SH2 (Src Homology-2)-SH3 adaptor NCK2 (NCK Adaptor Protein-2), thereby coupling Growth Factor signaling to Integrin signaling. Unlike ILK, PINCH does not contain a catalytic domain and therefore the primary role of PINCH is to mediate intermolecular interactions. The interaction between PINCH and NCK2 elevates the level of intracellular PIP3 through PI3K activation enhancing ILK signaling. PINCH binds to RSU1 (Ras Suppressor Protein-1) and T-Beta4 (Thymosin-Beta-4) to influence JNK (c-Jun Kinase) signaling and cell migration/survival/adhesion (G-Actin polymerization), respectively ILK acts upstream to JNK signaling and perturb the stability of the Tight Junction barrier (barrier dysfunction) resulting in the loss of Ocln (Occludin). Cytokines like TNF-Alpha (Tumor Necrosis Factor-Alpha)/TNFR (Tumor Necrosis Factor Receptor) activates the Integrin-ILK-GSK3 (Glycogen Synthase Kinase-3)-p130CAS (Crk-Associated Substrate-p130)-JNK signaling and regulate Ocln levels near junction points. TNF-Alpha induced activation of Rac1 modulates MAPK Signaling to activate JNKs in order to regulate AP-1 (Activator Protein-1)-mediated MMP (Matrix Metalloproteinase)-2/9 gene expression and tissue morphogenesis. ILK induced PINCH might regulate JNK signaling and the expression and/or stability of PINCH and RSU1 are mutually dependent on one another. However, the relative contributions of PINCH and RSU1 in modulating JNK signaling have not been determined. But here RSU1 is known to function as a negative regulator of Growth Factor induced JNK activation (Ref.2).
On Integrin engagement with ECM, PI3K is activated, through FAK (Focal Adhesion Kinase) and clustering and co-activation of Growth Factor/RTKs. ILK is then activated through PI3K, allowing binding with Alpha-Parvin and Pxn, and recruitment to focal adhesion plaques. At the focal adhesion plaques, ILK activity is crucial for maintaining upstream signaling to Beta1-Integrins and downstream signaling to Akt and GSK3Beta (Ref.6). The CH2 domain of Beta-Parvin bind to Alpha-Actinin, another Actin-binding protein, and this interaction depends on phosphorylation of the CH2 domain of Beta-Parvin by ILK. Pxn also interact with ILK and inhibiting ILK activity results in the loss of Pxn localization to focal adhesions. ILK phosphorylates the Beta-Integrin cytoplasmic domain at Thr788 (Threonine-788) and Thr789 of the Beta1-Integrin cytoplasmic domain, which are implicated in Integrin function. ILK activation stimulates phosphorylation of Akt, GSK3 and PHI1 (Phosphatase Holoenzyme Inhibitor-1), and both Akt and PDK-1 (Phosphoinositide Dependent Kinase-1) associate with ILK. Ser343 (Serine-343) within the activation loop of ILK is required for the interaction of PDK-1 with ILK (Ref.3). Binding of cells to ECM components via Beta1-Integrins stimulates ILK and downstream targets Akt (phosphorylation activation at amino-acid residues Ser473 and Thr308 (via PDK-1) and GSK3Beta (phosphorylation inhibition at amino-acid residue Ser9). These events suppress apoptosis and promote survival by inhibiting BAD (Bcl2-Antagonist of Cell Death), Caspase-3/9 and cell cycle transition by blocking proteolysis of CcnD1 (Cyclin-D1). Irradiation is able to activate this pathway, which then does not stimulate proliferation but rather blocks cells in the G2-Phase possibly allowing damage repair (Ref.7). Other downstream targets of ILK-induced Akt include mTOR (Mammalian Target of Rapamycin), NF-KappaB (Nuclear Factor-KappaB) and Alpha-NAC (Alpha-Nascent Polypeptide Associated Complex). mTOR activates HIF1Alpha (Hypoxia-Inducible Factor-1-Alpha Subunit) that along with NF-KappaB controls the gene expressions of iNOS (Inducible Nitric Oxide Synthase), COX2 (Cyclooxygenase-2) and VEGF (Vascular Endothelial Growth Factor); VEGF acts upstream to PI3K and in turn activates ILK. ILK is therefore implicated in the regulation of anchorage-dependent cell growth and survival, cell cycle progression EMT (Epithelial Mesenchymal Transition), invasion and migration, cell motility and contraction, vascular development (Vasculogenesis), Tubulo-Interstitial Fibrosis and Tumor Angiogenesis. Further ILK phosphorylation of GSK3Beta also inhibits Ctnn-Beta (Catenin-Beta) and LEF (Lymphoid Enhancer-Binding Factor)/TCF (Transcription Factor) interaction and in turn regulates function of Myc, Cadherins and CREB (cAMP Response Element-Binding Protein). ILK may directly activate CREB through the ERK (Extracellular Signal-Regulated Kinase)-MSK (Mitogen- and Stress-Activated Protein Kinase)-1/2 signaling route. ILK induced inhibition of GSK3 also alters c-Jun/AP-1 mediated gene transcription (Ref.2).
In addition, ILK-dependent pathway also activates SNAI2 (Snail Homolog-2)/Slug in colon carcinoma cells. Different pathways converge in SNAI to trigger EMT and this place SNAI in a central position in the process. EMT is the mechanism by which epithelial cells that are generated in a particular region dissociate from the epithelium and migrate to reach different locations. It is known that down-regulation of E-Cadherin/CdhE (Epithelial Cadherin) is essential for ingression of the mesodermal cells at Gastrulation and this is in agreement with SNAI/ZEB1 (Zinc Finger Homeodomain Enhancer-Binding Protein) acting as a repressor of E-Cadherin expression (Ref.1). SNAI/Slug must have additional targets that are independent of E-Cadherin. With regard to EMT, in addition to E-Cadherin, SNAI/Slug also down-regulate other epithelial markers, such as Dsp (Desmoplakin), the epithelial Muc1 (Mucin-1) and Krt18 (Keratin-18)/Cytokeratin-18. The epithelial markers are essential for regulation of junction assembly at desmosomes, regulation of intermediate filaments and cell adhesion. Mesenchymal markers such as Vim (Vimentin) and Fn (Fibronectin) are up-regulated and re-distributed; these regulate wound healing, cell adhesion, cell motility and Opsonization. SNAI/Slug is also responsible for the activation of RhoGTPases required for the modulation of cytoskeletal reorganization through the Rho Family GTPases signaling (Ref.8). Furthermore, the elucidation of the three-dimensional structure of ILK is critical to adequately address the functions of ILK as a kinase and as an adaptor molecule. ILK is an intracellular protein, which interacts with the cytoplasmic domains of Integrin Beta1 and Beta3 subunits (Ref.1 & 2). ILK regulates cell-ECM interactions, cytoskeletal organization and cell signaling with important, indispensable roles in various stages of embryonic development. ILK also has important roles in cancer progression, and has emerged as a valid therapeutic target in cancer. The recent findings of increased levels of ILK in various cancers, and that inhibition of ILK expression and activity is anti-tumorigenic, makes ILK an attractive target for cancer therapeutics (Ref.3). In terms of its role in cancer progression, the molecular basis for the high levels of ILK protein expression in various types of tumors is unclear and needs to be explored. ILK expression and activity are up-regulated in several types of cancers. Over-expression of ILK in vivo promotes Hyperplasia and Tumorigenesis. Inhibition of ILK activity in tumor cells reverses many of the malignant properties of tumor cells and as such ILK is a promising cancer therapeutic target (Ref.1). There is also a need to develop transgenic models for ILK dysregulation in multiple tissues to gain insight into the in vivo role of ILK in tumour progression and to identify tissues and cell types where targeting ILK dysregulation might be particularly effective. Further insights into the workings of this enigmatic protein might come from unexpected places, and might provide important information on how its dysregulation might be exploited to achieve therapeutic control of cancer (Ref.3).
References
- 1
- Persad S, Dedhar S. The role of integrin-linked kinase (ILK) in cancer progression.
- 2
- Legate KR, Montanez E, Kudlacek O, Fassler R. ILK, PINCH and parvin: the tIPP of integrin signalling.
- 3
- Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK.
- 4
- Sepulveda JL, Wu C. The parvins.
- 5
- Blattner SM, Kretzler M. Integrin-linked kinase in renal disease: connecting cell-matrix interaction to the cytoskeleton.
- 6
- Attwell S, Mills J, Troussard A, Wu C, Dedhar S. Integration of cell attachment, cytoskeletal localization, and signaling by integrin-linked kinase (ILK), CH-ILKBP, and the tumor suppressor PTEN.
- 7
- Cordes N, van Beuningen D. Cell adhesion to the extracellular matrix protein fibronectin modulates radiation-dependent G2 phase arrest involving integrin-linked kinase (ILK) and glycogen synthase kinase-3beta (GSK-3beta) in vitro.
- 8
- Nieto MA. The snail superfamily of zinc-finger transcription factors.
 关于我们
关于我们