IGF1R_Signaling
发布时间:2019-12-10 16:04 来源:SABiosciences
- 通路
- 概述
Review
Programmed cell death, a form of altruistic suicide is a genetically controlled means of cellular self-destruction that leads to dismantling and packaging of cell material for removal by phagocytosis. All cells possess the ability to undergo programmed cell death (otherwise known as apoptosis), and the process is essential for normal development to shape organs and tissues as well as to remove damaged cells. Although the cell may require de novo synthesis of some signaling molecules, the machinery for apoptosis is constantly present and may be rapidly activated. Therefore, the process of apoptosis needs tight regulation, and such regulation has been shown to be brought about by signaling through peptide growth factors including the IGF (Insulin-Like Growth Factor) system (Ref.1). The IGF system has a broad and potent ability to prevent apoptosis both during normal development and during stress or disease, in a wide array of cell types as diverse as osteoblasts, melanoma cells, cardiac myoblasts, neuronal cells, and epithelium. IGF1R (IGF1 Receptor), activated by its ligands, plays a pivotal role in tissue homeostasis, regulating cell proliferation, differentiation and migration during development and in the adults. Activation of the IGF1R is a particularly important survival-promoting signal (Ref.2).
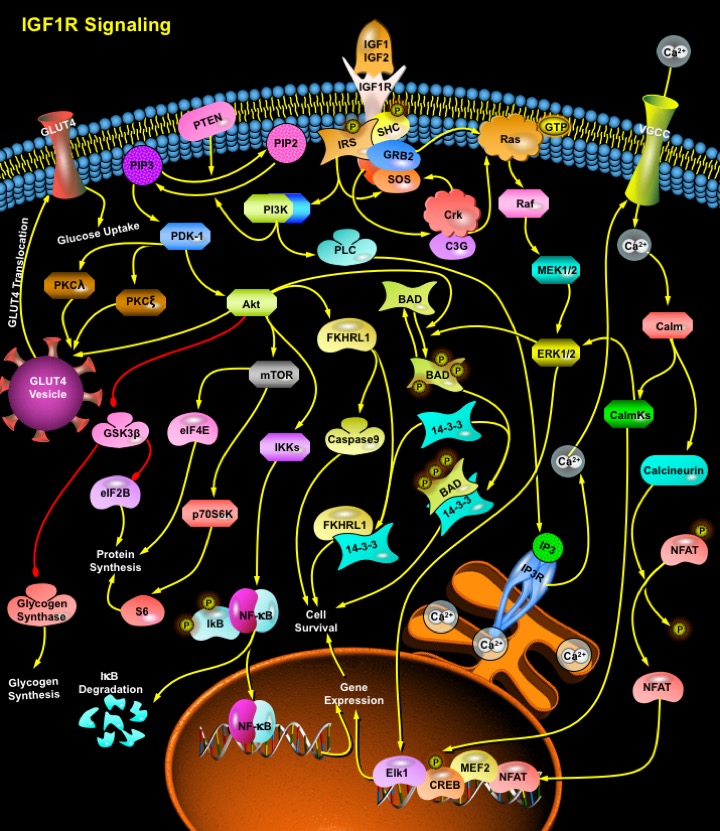
IGF1R is a transmembrane, ligand-activated tyrosine protein kinase that consists of Alpha2Beta2 heterotetramers held together by disulfide bridges. Both IGF1 and IGF2 exhibit high-affinity binding to IGF1R and most of the biological effects of IGF1 and IGF2 are mediated by IGF1R. The ligands bind to the cysteine-rich domain of the Alpha-subunits, leading to the transmission of a signal through the transmembrane domain to the Beta-subunit. The Beta-subunit responds by undergoing a conformational change that causes stimulation of tyrosine kinase activity, followed by autophosphorylation of a cluster of tyrosine residues of the IGF1R. Once these residues are phosphorylated, the intrinsic tyrosine kinase activity of the IGF1R increases, leading to phosphorylation of other sites in the receptor and associated substrate proteins. The main signaling pathway for IGF1R-mediated protection from apoptosis rests on the activation of PI3K (Phosphatidylinositol-3-Kinase) and Akt/PKB (Protein Kinase-B). The IGF1R also activates alternative pathways for protection from apoptosis and in some cases, involved in cell proliferation and differentiation. One of these pathways leads to the activation of MAPKs (Mitogen-Activated Protein Kinases), while a third pathway results in the Calcium mobilization inside the cell. All these pathways, however, result in maintenance of cell survival by antagonizing the processes and proteins involved in apoptosis. The multiplicity of signaling pathways used by the IGF1R may explain why this receptor has such a powerful and widespread antiapoptotic activity (Ref.2, 3 & 4).
The best-defined pathways by which IGF1R signaling can prevent apoptosis is mediated by PI3K. The binding of IGF1 or IGF2 to IGF1R activates the receptor's intrinsic tyrosine kinase activities, which results in the phosphorylation of the IRSs (Insulin Receptor Substrates) (Ref.5). Tyrosine-phosphorylated IRSs interact with the cytoplasmic protein PI3K through its SH2 (Src Homology -2) domains. PI3K activation then leads to the transduction of the functional effects of IGFs, such as enhanced glucose transport, enhanced cardiomyocyte contractility, and the inhibition of apoptosis by activating several downstream proteins and molecules. Activated PI3K catalyzes the conversion of PIP2 (Phosphatidylinositol 4,5-Bisphosphate) to PIP3 (Phosphatidylinositol 3,4,5-Trisphosphate), and the reverse reaction is catalyzed by PTEN (Phosphatase and Tensin Homolog). Elevated PIP3 binds to the PH (Pleckstrin Homology) domain of at least two proteins, Akt and PDK-1 (Phosphoinositide Dependent Kinase-1). PDK-1 then phosphorylates residue Thr308 on Akt. PDK-1 also phosphorylates PKC (Protein Kinase-C) proteins: PKC-Lambda (Protein Kinase-C-Lambda) and PKC-Zeta (Protein Kinase-C-Zeta) (Ref.4). These PKCs, along with Akt enhance the rate of Glucose uptake by the cell by facilitating the GLUT4 (Glucose Transporter-4) translocation from the GLUT4 vesicle to the membrane (Ref.6). Activated Akt phosphorylates and inactivates several proteins that are involved in apoptosis. A primary target is the BAD. In its non-phosphorylated state, BAD locates at the mitochondrial membrane where it interacts with BCL2 (B-Cell CLL/Lymphoma-2) and prevents it from performing its anti-apoptotic functions. Once phosphorylated by Akt, BAD associates with the cytosolic protein 14-3-3 and is unable to interfere with BCL2. Akt can also prevent the initiation of the Caspase cascade through phosphorylation and inactivation of Caspase9. Akt also phosphorylates several pro-apoptotic members of the forkhead transcription factor family, FKHRL1, FKHR and prevents their activity (Ref.7). Action of Akt diminishes expression of FasL (Fas Ligand), thus decreasing Fas-mediated apoptosis. In addition to the inhibition of pro-apoptotic transcription factors, the activity of Akt also increases the levels of anti-apoptotic proteins including BCL2 and BCL-X and several extracellular matrix adhesion molecules. Induced activity of Akt also leads to expression of the anti-apoptotic transcription factor NF-KappaB through regulation of IKKs (I-KappaB Kinases). This results in I-KappaB degradation and allows NF-KappaB to enter the nucleus and activate transcription of anti-apoptotic genes. A further protective pathway activated by Akt involves inhibition of GSK3 (Glycogen Synthase Kinase-3) which results from its phosphorylation at an N-terminal serine residue (Ser21 in GSK3Alpha and Ser9 in GSK3Beta) (Ref.6). In response to IGFs, the inhibition of GSK3 promotes the dephosphorylation and activation of Glycogen Synthase, contributing to the stimulation of glycogen synthesis. GSK3 also catalyses the phosphorylation and inhibition of eIF2B (Eukaryotic Protein Synthesis Initiation Factor-2B), thereby inhibiting protein synthesis. Hence IGF1R, by inhibiting GSK3, stimulates the dephosphorylation and activation of eIF2B, contributing to an increased rate of protein synthesis. IGF1 promotes protein synthesis by activating eIF4E (Eukaryotic Initiation Factor-4E). In the resting state, eIF4E is held in an inactive complex by one of its three binding proteins: 4EBP1, 4EBP2, or 4EBP3. Phosphorylation of 4EBP results in dissolution of the eIF4E-4EBP complex, freeing eIF4E to promote the initiation phase of protein translation. The mTOR is also a phosphorylation substrate for Akt. Phosphorylated mTOR promotes phosphorylation and inhibition of 4EBP1, and promotes protein synthesis by relieving 4EBP-mediated inhibition of eIF4E. When phosphorylated by mTOR, the ribosomal p70S6K and S6 (Ribosomal Protein-S6) becomes activated and increases protein synthesis (Ref.8).
Upon IGF1R autophosphorylation the protein SHC (SH2 Containing Protein) is recruited to the receptor and becomes phosphorylated on tyrosine residues. Activated SHC then binds the adaptor GRB2 (Growth Factor Receptor Bound Protein-2), recruiting the SOS in an IRS-independent manner. This complex then activates Ras and initiates sequential phosphorylation cascades involving serine/threonine kinase Raf, MEK1/2 (MAP Kinase Kinases), and ERK1/2 (Extracellular Signal Regulated Kinases). This pathway of IGF1R signaling has been most closely associated with cell differentiation and migration, but in some cases also can regulate the machinery of apoptosis, for example, in detachment-induced death, or Anoikis. An endpoint of the MAPK pathway is modification of transcription factor activity, such as activation of Elk transcription factors. SRF (Serum Response Factor) and Elk1 contribute to mitogenic signaling by many factors. Similar to the Akt pathway, the downstream target of ERKs that prevents apoptosis is BAD. However, JNK promotes apoptosis and the role of IGF1R signaling is to prevent the activation of JNK. The Ras-->Raf-->ERK1/2 pathway can also be activated by the tyrosine phosphorylation of IRSs, with the resultant formation of the IRS-GRB2-SOS complex that activates Ras, which in turn binds to and activates Raf, subsequently phosphorylating and activating MEKs, followed by ERKs. Phosphorylated ERKs, in turn, transmit signals to the nucleus, with resultant mitogenic response: progression of the cell cycle and cell proliferation (Ref.6 & 7).
In addition to modulation of intermediate regulators of cell survival through activation of enzymatic cascades, the IGF1R also can modify Calcium-dependent signaling pathways. IGF1R acts as modulator of ion channels. Ligand binding to IGF1R leads to activation of voltage-dependent Calcium channels; thus, causing large transient increases in the Ca2+ levels. The downstream effects include regulation of Calcium-dependent transcription factors such as MEF2 (Mads Box Transcription Enhancer Factor-2), NFAT (Nuclear Factors of Activated T-cells) and CREB. These transcription factors promote the expression of several anti-apoptotic proteins including BCL2. The increased Ca2+ levels in the cytoplasm activate the protein phosphatase Calcineurin by disrupting the inhibitory effects of Calm (Calmodulin). Calcineurin activation leads to the dephosphorylation of NFAT, allowing it to enter the nucleus, where it cooperates with other transcription factors to bind promoters. The CalmKs (Ca2+/Calm [Calmodulin]-dependent Protein Kinases), activated by the the increased Ca2+ levels activate the transcription factor CREB, and has also been suggested to activate the ERK1/2 pathway, ultimately promoting the process of IGF1R-induced cell survival (Ref.4, 5 & 6).
IGF1R signalling plays numerous roles in both physiological and pathophysiological conditions. Under normal physiological conditions IGF-1 enhances DNA and protein synthesis in cardiomyocytes, it promotes myofibril development, and it is necessary for entry into the S-phase of the cell cycle. IGF1 also has an important role in the development of the hypertrophic response, where it enhances the expression of contractile proteins such as actin, myosin and troponin. Through its tyrosine kinase, PI3K and MAPK signalling pathways, IGF1R may also reduce the risk of heart failure through the prevention of apoptosis. IGFs protect cells from apoptosis induced by a wide variety of conditions, including growth factor withdrawal, chemotherapy and oncogene expression. Activation of IGF1R by its ligand also initiates metabolic cascades that result in the stimulation of protein synthesis, glucose intake, glycogen synthesis, and lipid storage (Ref.8). Since the ability to regulate apoptosis may impact upon several types of serious diseases in humans, including different human Cancers, drugs to disrupt IGF1R function have been developed and are now entering clinical trial. Understanding of the cellular pathways by IGF1R that signal to a cell to live or die is crucially important for the development of appropriate clinical treatments (Ref.9 & 10).
References
- 1
- Vincent AM, Feldman EL. Control of cell survival by IGF signaling pathways.
- 2
- Torres Aleman I. Role of insulin-like growth factors in neuronal plasticity and neuroprotection.
- 3
- Shelton JG, Steelman LS, White ER, McCubrey JA. Synergy between PI3K/Akt and Raf/MEK/ERK pathways in IGF-1R mediated cell cycle progression and prevention of apoptosis in hematopoietic cells.
- 4
- Kuemmerle JF. IGF-I elicits growth of human intestinal smooth muscle cells by activation of PI3K, PDK-1, and p70S6 kinase.
- 5
- Tseng YH, Ueki K, Kriauciunas KM, Kahn CR. Differential roles of insulin receptor substrates in the anti-apoptotic function of insulin-like growth factor-1 and insulin.
- 6
- Mora A, Sakamoto K, McManus EJ, Alessi DR. Role of the PDK1-PKB-GSK3 pathway in regulating glycogen synthase and glucose uptake in the heart.
- 7
- Zheng WH, Kar S, Quirion R. Insulin-like growth factor-1-induced phosphorylation of transcription factor FKHRL1 is mediated by phosphatidylinositol 3-kinase/Akt kinase and role of this pathway in insulin-like growth factor-1-induced survival of cultu
- 8
- Senthil D, Choudhury GG, Abboud HE, Sonenberg N, Kasinath BS Regulation of protein synthesis by IGF-I in proximal tubular epithelial cells.
- 9
- Phan TT, Lim IJ, Bay BH, Qi R, Longaker MT, Lee ST, Huynh H. Role of IGF system of mitogens in the induction of fibroblast proliferation by keloid-derived keratinocytes in vitro.
- 10
- Miller BS, Yee D. Type I insulin-like growth factor receptor as a therapeutic target in cancer.
 关于我们
关于我们