Fas_Signaling
发布时间:2019-12-10 11:38 来源:SABiosciences
- 通路
- 概述
Review
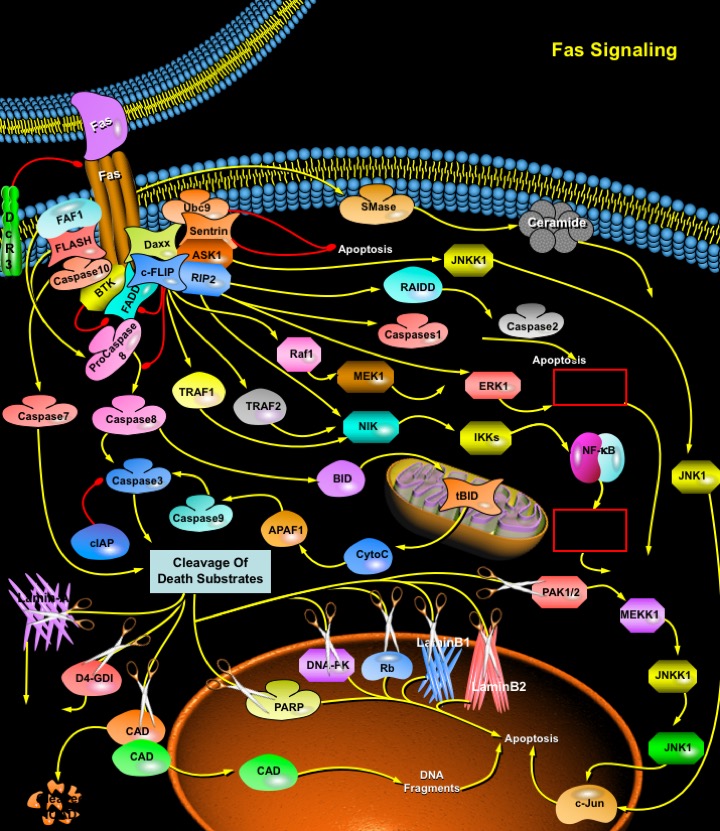
Fas (also called Apo1 or CD95) is a death domain-containing member of the TNFR (Tumor Necrosis Factor Receptor) superfamily. It has a central role in the physiological regulation of Programmed Cell Death and has been implicated in the pathogenesis of various malignancies and diseases of the immune system. Although the FasL (Fas Ligand)-Fas system has been appreciated mainly with respect to its death-inducing function, it also transduces proliferative and activating signals through pathways that are still poorly defined. The Fas Receptor induces an apoptotic signal by binding to FasL expressed on the surface of other cells. Fas is a Type-I transmembrane protein, where as FasL a Type-II Transmembrane protein of TNF family and can be shed in a soluble form by action of metalloproteinase (Ref.1).
The Fas Receptor upon binding to the FasL trimerizes and induces Apoptosis through a cytoplasmic domain called DD (Death Domain) that interacts with signaling adaptors like FAF-1 (Fas-Associated Factor-1), FADD (Fas-Associated Death Domain), Daxx, FAP-1, FLASH (FLICE-associated huge) and RIP (Receptor-Interacting Protein). FADD carries a DED (Death Effector Domain) and by homologous interaction it recruits the DED containing Procaspase8 protein which is in inactive state. This protein complex is also known as DISC (Death-Inducing Signaling Pathways) and exists in Type-I cells. Procaspase8 is proteolytically activated to Caspase8. FADD also helps in the activation of Caspase10. Upon activation, Caspase8 and Caspase10 cleave and activate downstream effector Caspases, including Caspase3, 6 and 7. Activated Caspase8 activates Caspase3 through two pathways; The complex one is that Caspase8 cleaves BID (Bcl2 Interacting Protein) and its COOH-terminal part translocates to mitochondria where it triggers the release of mitochondrial pro-apoptotic factors like CytoC (Cytochrome-C) and SMAC (Second Mitochondria-derived Activator of Caspases) also called Diablo. The released CytoC binds to APAF1 (Apoplectic Protease Activating Factor-1) together with dATP and Procaspase9 and activates Caspase9. Caspase9 cleaves Procaspase3 and activates Caspase3. Another pathway is that Caspase8 cleaves Procaspase3 directly and activates it. Both pathways are regulated at the level of Caspase8 activation by the endogenous inhibitor FLIP (FLICE (FADD Like IL-1Beta-Converting Enzyme)-Inhibitory Protein), which may also be recruited by FADD. Interestingly, FLIP may also participate in an alternate signalling pathway, recruiting TRAF1 (Tumor Necrosis Factor-Associated Factor-1), TRAF2 (Tumor Necrosis Factor-Associated Factor-1), the MAPKKK (MAP Kinase Kinase Kinase) Raf1 and RIP to activate ERK (Extracellular signal-Regulated Kinase) and NF-KappaB (Nuclear Factor-KappaB) pathways, leading to proliferation and/or inflammation. This differential activity of FLIP, which appears to reflect activity of short vs long FLIP isoforms that promote the death vs proliferation pathways, respectively, mediates a major decision in response to Fas signalling: death vs proliferation/inflammation. Caspases are inhibited by IAPs (Inhibitor of Apoptosis Proteins) (Ref.2 & 3). Caspase3, 6 and 7 once activated lead to breakdown of several cytoskeletal and nuclear proteins (structural, signaling proteins or kinases) like GDID4 (GDP-Dissociation Inhibitor-D4), PARP (Poly ADP-Ribose Polymerase), Alpha-Fodrin, GAS2 (Growth Arrest Specific-2) Lamin-A and B (B1 and B2) and PAK (p21-Activated Kinase) thus inducing apoptosis. PAK activity resulting from Caspase-mediated cleavage is a necessary component of JNK (c-Jun Terminal Kinase) activation induced by Fas receptor signaling and thus PAK can also contribute to the induction of cell death. Caspase3 also cleaves ICAD, the inhibitor of CAD (Caspase Activated DNAse), which frees CAD to enter the nucleus and cleave DNA. In the nucleus, activated Caspase3 also cleaves DNA-PKcs and release the cleaved fragments in the cytosolic compartment. Stimulation of Fas also results in significant alterations of Rb (Retinoblastoma protein) (Ref.4 & 5).
Besides the FADD/Caspase8 signaling cascade, a number of other signaling pathways are also activated by the Fas Receptor. Among the molecules involved in Fas-mediated signaling, Daxx and RIP have been shown to bind to the FasR, modulating its signal. Daxx (Death-Domain Associated protein) binds to the Fas death domain although Daxx itself does not contain a DD. Daxx and FADD bind independently to Fas and activate distinct pathways. Daxx can enhance Fas-mediated apoptosis by activating the JNK kinase cascade, culminating in the phosphorylation and activation of transcription factors such as c-Jun. The JNK kinase kinase ASK1 (Apoptosis Signal Regulating Kinase 1) sequesters Daxx in the cytoplasm. After Fas stimulation, Daxx interacts with and activates ASK1. In the absence of ASK1, Daxx is found in the nucleus, where it localizes to PODs (PML (Promyelocytic Leukemia Protein) Oncogenic Domains). ASK1 and Daxx can induce a Caspase-independent form of cell death that is independent from the kinase activity of ASK1. ASK1 is also able to induce Caspase-dependent apoptosis under critical requirement of its kinase activity. Daxx also interact with Sentrin, a ubiquitin-like protein that can covalently modify cellular proteins, and is Fas binding protein that protects cells against Fas induced cell death. Also, Daxx interacted with Ubc9, an essential protein as a key-conjugating enzyme. Colocalization of Fas, Sentrin, and Ubc9 binding regions suggests the importance of that region upon the regulation of Daxx. In addition, FLASH enhances the activation of Caspase8 in Fas-mediated apoptosis. Thus, DED-containing proteins seem to modulate the apoptotic process. FAF1 is a Fas-associating molecule, which enhances Fas mediated apoptosis. The N-terminus of FAF1 binds to the DD of Fas even though it does not contain the typical death domain. FAF1 is a component of Fas-DISC, and DISC is formed by interaction of the DED-like region (amino acid 181–381 of FAF1) of FAF1 and the DEDs of Caspase8 and FADD (Ref.6, 7 & 8).
The Receptor Interacting Proteins (RIP, RIP2, RIP3, RIP4) are death domain-containing proteins which, uniquely, possess serine/threonine kinase activity. RIP associates with the Fas death domain but can also interact directly with FADD and FLIP. RIP overexpression results in NF-KappaB translocation, JNK activation, and apoptosis. RIP family proteins may transduce non-apoptotic signal through several pathways, including NF-KappaB and ERK. NF-KappaB activation by RIP is dependent on functional NIK (NF-KappaB-Inducing Kinase), and may involve NIK recruitment into the DISC. RIP mediated activation of NF-KappaB, while RIP is bound to FLIP, may account at least in part for the observation that FLIP can regulate NF-KappaB activation. Activation of NF-KappaB results in its translocation to the nucleus, where it acts as a transcription factor. NF-KappaB activation usually induces proliferation, differentiation, or inflammation, but may also promote apoptosis, perhaps accounting for the discrepancies in reported RIP functions in different cellular contexts. Activated RIP2 can phosphorylate ERKs (Extracellular signal-Regulated Kinases), thus activating the ERK pathway independently of MEK. Finally, RIP can also activate Caspase1 directly, and Caspase2 via the adaptor protein RAIDD (RIP-associated ICH-1/Ced3-homologous protein with a Death Domain). Activation of Caspase1 allows the processing of IL-1Beta (pro-Interleukin-1Beta) to active IL-1Beta, a potent inducer of inflammation. Thus, RIP can mediate Fas-induced differentiation and inflammation through a number of different pathways. An acidic SMase (Sphingomyelinase) is also activated in response to stimulation of Fas, resulting in the production of Ceramide, a mediator of cell stress and well known stimuli of apoptosis (Ref.2 & 9).
In B-Cells, BTK (Bruton's Tyrosine Kinase) associates with the death receptor Fas and impairs its interaction with FADD, which is essential for the recruitment and activation of FLICE by Fas during the apoptotic signal, thereby preventing the assembly of DISC after Fas-ligation. Fas pathway can also be regulated at the transcriptional level, such as by the tumour suppressor p53. p53 positively regulates genes, including Fas and Bax that are involved in FasL-induced apoptosis. FasL has been implicated in maintaining immune privileged sites such as the eye, testis, brain, joints, and pregnant uterus by inducing apoptosis in activated infiltrating leukocytes and lymphocytes that express Fas. Since, Fas and FasL are known regulators of apoptosis in cells of the immune system, blocking Fas/FasL interactions in human colon carcinoma cells can lead to thymine less death. Fas/FasL interaction may also be involved in target destruction during organ-specific autoimmune diseases, such as Hashimoto's Thyroiditis, Insulin-dependent Diabetes Mellitus and Multiple Sclerosis (Ref.10 & 11).
References
- 1
- Lopez-Hernandez FJ, Ortiz MA, Piedrafita FJ. The extrinsic and intrinsic apoptotic pathways are differentially affected by temperature upstream of mitochondrial damage.
- 2
- Mollinedo F, Gajate C. Fas/CD95 death receptor and lipid rafts: New targets for apoptosis-directed cancer therapy.
- 3
- Luschen S, Falk M, Scherer G, Ussat S, Paulsen M, Adam-Klages S. The Fas-associated death domain protein/caspase-8/c-FLIP signaling pathway is involved in TNF-induced activation of ERK.
- 4
- Kwon KY, Jang JH, Choi WI, Ramachandran S, Cho CH, Cagle PT. Expression of apoptotic nuclei by ultrastructural terminal deoxyribonucleotidyl transferase mediated dUTP nick end labeling and detection of FasL, caspases and PARP protein molecules in cad
- 5
- Sikora E, Bielak-Zmijewska A, Magalska A, Piwocka K, Mosieniak G, Kalinowska M, Widlak P, Cymerman IA, Bujnicki JM. Curcumin induces caspase-3-dependent apoptotic pathway but inhibits DNA fragmentation factor 40/caspase-activated DNase endonuclease i
- 6
- Matsuyoshi S, Shimada K, Nakamura M, Ishida E, Konishi N. FADD phosphorylation is critical for cell cycle regulation in breast cancer cells.
- 7
- Salomoni P, Khelifi AF. Daxx: death or survival protein?
- 8
- Bernardi R, Pandolfi PP. Role of PML and the PML-nuclear body in the control of programmed cell death.
- 9
- Lavrik IN, Golks A, Baumann S, Krammer PH. Caspase-2 is activated at the CD95 Death-Inducing Signaling complex in the course of CD95-induced apoptosis.
- 10
- Lin HL, Chen CJ, Tsai WC, Yen JH, Liu HW. In vitro folate deficiency induces apoptosis by a p53, Fas (Apo-1, CD95) independent, bcl-2 related mechanism in phytohaemagglutinin-stimulated human peripheral blood lymphocytes.
 关于我们
关于我们