FAK1_Signaling
发布时间:2019-12-10 11:23 来源:SABiosciences
- 通路
- 概述
Review
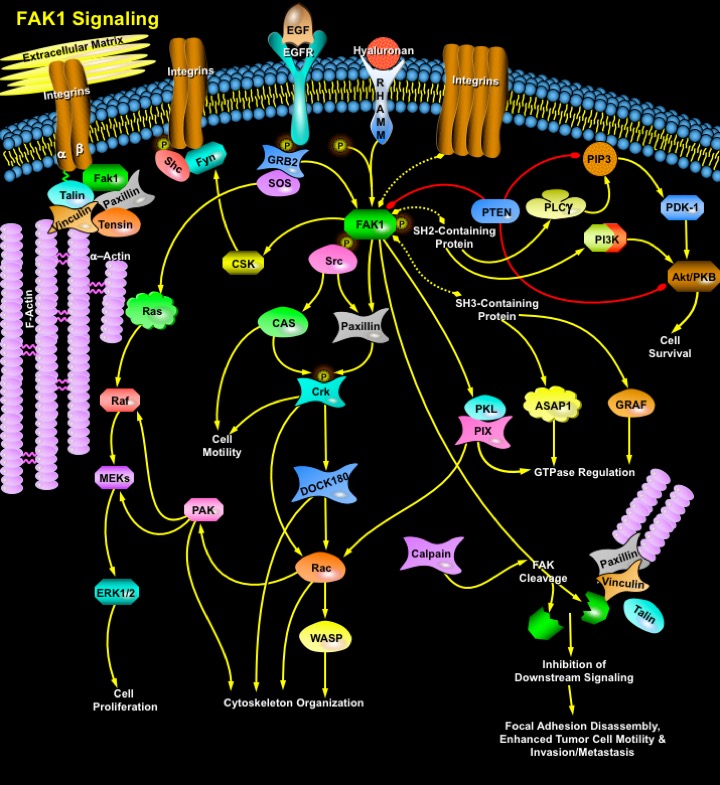
Engagement of integrin receptors with extracellular ligands gives rise to the formation of complex multi-protein structures that link the ECM (Extracellular Matrix) to the cytoplasmic actin cytoskeleton and signaling proteins including Talin, Alpha-actinin, Vinculin, Zyxin, Paxillin and FAK (Focal Adhesion Kinase). These adhesive complexes are dynamic, often heterogeneous structures, varying in size and organization, and signaling through these complexes and focal adhesions have been implicated in the regulation of a number of key cellular processes, including growth factor induced mitogenic signals, cell survival, cell proliferation and migration, cell locomotion and regulation of cell cycle (Ref.1). FAK is a non-receptor cytosolic PTK with a central catalytic domain flanked by large N- and C-terminal domains that indirectly localizes to sites of integrin-receptor clustering through interations with integrin-associated proteins (Ref.2). FAK can be subdivided into three domains; an amino-terminal domain that bind to the cytoplasmic tail of Beta-Integrin subunits and the intracellular domain of the EGFR (Epidermal Growth Factor Receptor); a central TK (Tyrosine Kinase) domain; and a carboxy-terminal domain that consists of two proline-rich motifs and a FAT (Focal Adhesion Targeting Sequence) that associate with other focal-adhesion proteins, including Paxillin and Talin (Ref.3).
Clustering of integrins leads to the rapid recruitment of FAK to the focal adhesion complex and its concurrent phosphorylation on tyrosine. Activated FAK is phosphorylated at Tyr397, which correlates with increased catalytic activity of FAK and is important for the tyrosine phosphorylation of focal complex associated proteins such as Paxillin and CAS. The phosphorylation on Tyr397 also creates a high affinity binding site recognized by the SH2 domain of Src family kinases and leads to the recruitment and activation of Src through the formation of a bipartite kinase complex. Activation of the FAK-Src complex is central to regulation of down-stream signaling pathways that control cell spreading, cell movement and cell survival (Ref.2 & 3). Phosphorylation of FAK is also important for the recruitment of other SH2-containing proteins, including PI3K Pphosphotidylinositol 3-Kinase) and PLC-Gamma (Phospholipase-C). As a result of its tyrosine phosphorylation, FAK forms a complex with an adapter protein GRB2 (Growth Factor Receptor-Bound Protein-2) that binds to the GDP/GTP exchange protein SOS. This complex activates Ras and the downstream mediators such as PI3K (Phosphatidylinositol-3 Kinase), MEKs (MAPK/ERK Kinases) and ERK (Extracellular Signal Regulated Kinase)/MAPK (Mitogen Activated Protein Kinase) cascades (Ref.4). Once activated by FAK or SHC (SH2 Containing Protein)-Fyn, Ras activates PI3K and Raf to the cytoplasmic membrane. In addition, FAK serves as a scaffold for SH3 domain-containing proteins like p130CAS (Crk-Associated Substrate), ASAP1 or GRAF (GTPase Regulator Associated with FAK), which bind to either of two C-terminal proline-rich regions (Ref.2). Phosphorylated CAS binds to Crk and DOCK180 (Dedicator of Cytokinesis-180), leading to an increase in the affinity of the membranes for Rac. Activated Rac regulate numerous biochemical pathways, including activation of MAPK/ERK, PIX (PAK-Interacting Exchange Factor), PAK (p21-Activated Kinase), PKL (Paxillin Kinase Linker) and p38/JNK (c-Jun N-terminal Kinase), the key regulators of gene expression and cell cycle (Ref.5). In addition, Rac activation is closely coupled to activation of WASP (Wiskott-Aldrich Syndrome Protein) allowing for the coincident and coordinated cycloskeleton organization. When Hyaluronan binds to RHAMM (Hyaluronan-Mediated Motility Receptor) the phosphorylation of FAK occurs, which is a necessary step for disassembly of focal contacts and subsequent motility. FAK is initially cleaved by Calpain into an amino-terminal fragment and a carboxy-terminal fragment. Calpain-mediated cleavage of FAK causes the dissociation of the FAT sequence and abolishes the kinase activity of FAK at adhesion sites, which impairs the ability of FAK to act as an adaptor molecule. This contributes to the disassembly of focal-adhesion structures and results in the loss of integrin, growth factor and Src-signaling to downstream effectors (Ref.3). Dephosphorylation of FAK also involves PTEN, but PTEN's predominant enzymatic activity appears to be the dephosphorylation of phosphoinositides, PI(3,4,5)P3 (Phosphatidylinositol 3,4,5-Trisphosphate) and PI(3,4)P2 (Phosphatidylinositol 3,4-Bisphosphate), thus antagonizing the downstream effectors, such as Akt, PDK-1 (Phosphoinositide Dependent Kinase-1) (Ref.4).
FAK plays a pivotal role in signal transduction at integrin-linked cellular adhesions, which mediate cell contact with the ECM. It plays a central role in the survival of anchorage-dependent cells and is essential for integrin-linked cell migration– the processes that play important roles in the development of malignancies. FAK is upregulated in a wide variety of human epithelial cancers, with expression being closely correlated to invasive potential. Recently, FAK expression has been implicated in either the progression of tumor cells to malignancy or the pathogenesis of cancer, raising the possibility that intervention strategies to block FAK function may potentially provide an opportunity for the development of anticancer therapeutics (Ref.6).
References
- 1
- Sawai H, Okada Y, Funahashi H, Matsuo Y, Takahashi H, Takeyama H, Manabe T. Activation of focal adhesion kinase enhances the adhesion and invasion of pancreatic cancer cells via extracellular signal-regulated kinase-1/2 signaling pathway activation.
- 2
- Caron-Lormier G, Berry H. Amplification and oscillations in the FAK/Src kinase system during integrin signaling
- 3
- Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology
- 4
- Yano H, Mazaki Y, Kurokawa K, Hanks SK, Matsuda M, Sabe H. Roles played by a subset of integrin signaling molecules in cadherin-based cell-cell adhesion.
- 5
- Wade R, Vande Pol S. Minimal features of paxillin that are required for the tyrosine phosphorylation of focal adhesion kinase.
- 6
- McLean GW, Avizienyte E, Frame MC. Focal adhesion kinase as a potential target in oncology.
 关于我们
关于我们