eNOS_Signaling
发布时间:2019-12-10 10:18 来源:SABiosciences
- 通路
- 概述
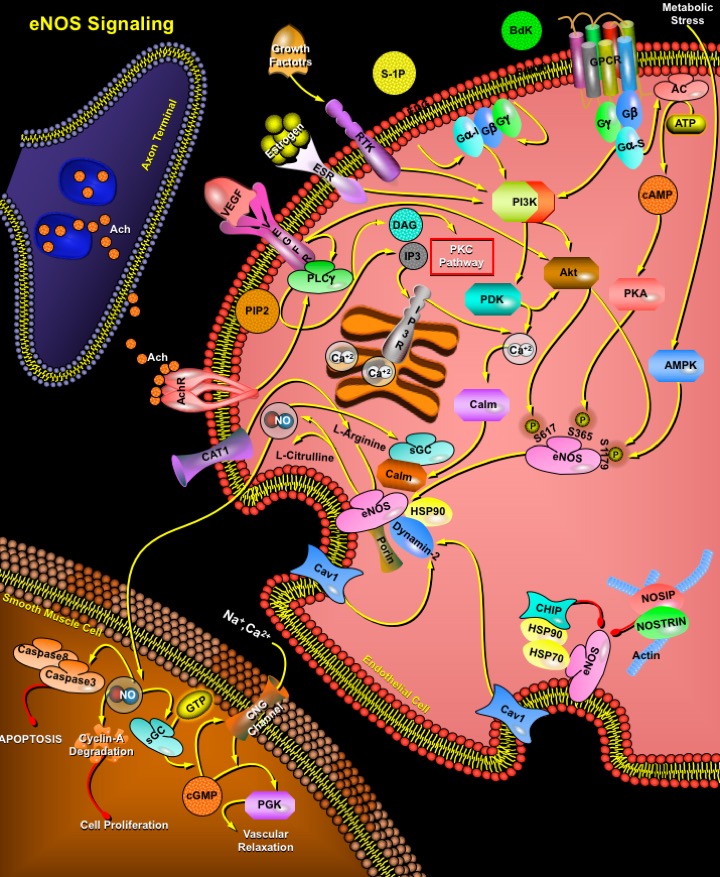
Review
NO (Nitric Oxide) is a short-lived free radical gas involved in diverse physiological and pathological processes. It is produced along with L-Citrulline by the oxidation of L-Arginine and catalyzed by three different isoforms of NOS (NO Synthase). Type-I nNOS (neuronal NOS) and Type-III eNOS (endothelial NOS) are constitutively expressed as latent enzymes and require a higher concentration of Ca2+ for the enzyme activity. In contrast, Type-II iNOS (inducible NOS) is Ca2+ independent because it’s high affinity for Ca2+/Calm (Calmodulin) renders the enzyme active even at basal levels of intracellular Ca2+ (Ref.1 & 2). The catalysis of this reaction requires a number of essential cofactors such as mononucleotide, FAD (Flavin Adenine Dinucleotide), and NADPH (Nicotinamide Adenine Dinucleotide, Reduced). The NO thus generated exerts a number of functions on the cardiovascular system.
NO production from endothelial cells is stimulated by a variety of mechanical forces such as shear stress and cyclic strain and humoral factors ranging from growth factors to peptide hormones, including Ach (Acetylcholine), VEGF (Vascular Endothelial Growth Factor), Bdk (Bradykinin), Estrogen, S-1P (Sphingosine-1Phosphate), H2O2 (Hydrogen Peroxide), and Angiotensin-II. eNOS is a dually acylated peripheral membrane protein that is targeted to endothelial plasmalemmal caveolae through an interaction with the caveolae structural protein, Cav1 (Caveolin-1). Cav1 inhibition of eNOS is relieved by Calm, which causes dissociation of eNOS from caveolin. This regulatory mechanism is further modified by HSP90 (Heat Shock Protein-90), which binds to eNOS and facilitates displacement of Cav1 by Calm. In addition to these protein interactions that modulate Calm binding, other cellular signaling cascades also regulate eNOS activity. Physiologically, endothelial cells are exposed to the hemodynamic forces of blood including laminar shear stress. Shear stress, via G-proteins (Gs) activate several signal transduction pathways, including the PI3K (Phosphatidylinositol 3-Kinase), PDK (Phosphoinositide-Dependent Kinase) and AC (Adenylate Cyclase) via cAMP (cyclic Adenosine Monophosphate), leading to eNOS activation by phosphorylation of serine residues (S617 and S1179 for Akt, and S635 and S1179 for PKA), which promote eNOS activation. Additional stimuli, such as VEGF, Estrogen, S-1P and Bdk (Bradykinin), bind to their cognate receptors (RTKs, VEGFR, ESR, EDG and BdkR) and stimulate PI3K/Akt. However, they also activate PLC-Gamma (Phospholipase-C) and PIP2 (Phosphatidyl Inositol 4, 5-Bisphosphate) to increase cytoplasmic calcium and DAG (Diacylglycerol) levels. The increase in cytoplasmic calcium levels activates Calm, which binds to the canonical Calm-binding domain in eNOS to promote the alignment of the oxygenase and reductase domains of eNOS, leading to efficient NO synthesis. In addition, Calm activates CalmK-II (Calm Kinase-II), which phosphorylates eNOS on S1179. Increases in DAG levels activate PKC (Protein Kinase-C) pathway, which may negatively regulate eNOS or influence its coupling. Finally, metabolic stress triggering the breakdown of ATP stimulates AMPK (AMP Kinase) to phosphorylate eNOS on S1179. Phosphorylation of this residue by PKA (Protein Kinase-A) is also associated with increased enzyme activity. Other proteins which are associated with increased eNOS activity or NO release are Dynamin-2 (a GTP-binding protein) and Porin, which colocalize and directly interact with eNOS. Their interactions with eNOS are stimulated by intracellular Ca2+ and lead to eNOS activation (Ref.3). Efficient supply with substrate during all this is ensured by localization of the arginine transporter CAT1 (Cationic Amino Acid Transporter-1) in caveolae and its direct interaction with eNOS.
eNOS can interact with various proteins in its `less active' and `more active' states. Myristoylation of eNOS occurs co-translationally and targets eNOS to cellular membranes, where eNOS is then palmitoylated. These lipidation promote eNOS association with cell membranes and are essential for linking upstream signal transduction pathways to eNOS activity in cells. N-myristoylated and palmitoylated membrane-bound eNOS associates with the caveolae coat protein Cav1 and with HSP90. CHIP (C-terminal HSP70-Interacting Protein) interacts with both HSP70 and HSP90, and negatively regulates eNOS trafficking into the Golgi complex. By contrast, NOSIP (Nitric Oxide Synthase-Interacting Protein), a 34-kDa protein and NOSTRIN (Nitric Oxide Synthase Traffic Inducer) negatively regulates eNOS localization in the plasma membrane. Acute activation of eNOS in blood vessels in response to agonist such as Ach (Acetylcholine) or Bdk results in the activation of the sGC (soluble Guanylyl Cyclase) in smooth muscle cells, production of cGMP (cyclic Guanosine Monophosphate) and degradation of Cyclin-A. An increase in intracellular cGMP levels affect vascular tone by decreasing the intracellular concentration of free [Ca2+]I by CNG channel (Cyclic-Nucleotide Gated Ion Channel Complex) modulation; as well as by activating PGK (Protein Kinase-G) and phosphorylating HSP20, which regulate force by binding to thin filaments and inhibit cross-bridge cycling (Ref.4). Nitrosylation of Caspase3 and Caspase8 inactivates the proteins, thus leading to inhibition of apoptosis.
eNOS is an important regulator of cardiovascular homeostasis because it is the major source of NO production in vascular endothelial cells. eNOS plays a crucial role in the state of blood vessel vasodilatation and hence blood pressure regulation. In addition, NO released from the endothelium modulates other processes including platelet aggregation, platelet and leukocyte adhesion to the endothelium, Endothelin-1 generation, vascular smooth muscle cell proliferation, and angiogenesis. Because of the important role of NO in each of these processes, abnormalities in vascular NO production are thought to contribute to the pathogenesis of certain vascular disorders such as those of atherosclerosis and hypertension (Ref.5 & 6).
References
- 1
- Petruson K, Stalfors J, Jacobsson KE, Ny L, Petruson B. Nitric oxide production in the sphenoidal sinus by the inducible and constitutive isozymes of nitric oxide synthase.
- 2
- Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase.
- 3
- Ostrom RS, Bundey RA, Insel PA. Nitric oxide inhibition of adenylyl cyclase type 6 activity is dependent upon lipid rafts and caveolin signaling complexes.
- 4
- Minshall RD, Sessa WC, Stan RV, Anderson RG, Malik AB. Caveolin regulation of endothelial function.
- 5
- Ho FM, Lin WW, Chen BC, Chao CM, Yang CR, Lin LY, Lai CC, Liu SH, Liau CS. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-kappaB and c-Jun NH(2)-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway
- 6
- Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases.
 关于我们
关于我们