Akt_Signaling
发布时间:2019-12-09 18:00 来源:SABiosciences
- 通路
- 概述
Review
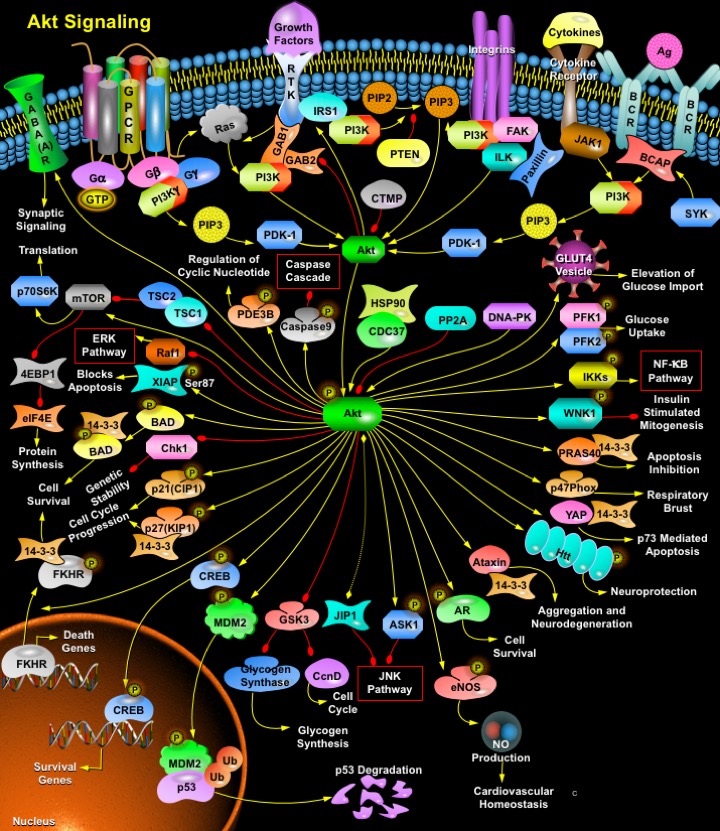
Akt (v-Akt Murine Thymoma Viral Oncogene)/ PKB (Protein Kinase-B) is a Serine/threonine Kinase that is involved in mediating various biological responses, such as inhibition of Apoptosis and stimulation of cell proliferation. Three mammalian isoforms are currently known: Akt1/PKB- Alpha, Akt2/PKB-Beta and Akt3/PKB-Gamma. All three isoforms of Akt share a common structure of three domains. The N-terminus of the protein is a PH (Pleckstrin Homology) domain, which interacts with membrane lipid products such as PIP2 (Phosphatidylinositol-3,4-Bisphosphate) and PIP3 (Phosphatidylinositol-3,4,5-Triphosphate). The PH domain is approximately 100 amino acids and plays a role in recognition by upstream kinases and membrane translocation of Akt. The center region of the protein is the Kinase domain, which has high similarity to other kinases. This domain contains a conserved threonine residue, which needs to be phosphorylated in order to activate Akt. The approximately 40 amino acids at the C-terminus of the protein form a regulatory domain that contains a proline rich region and a hydrophobic motif with a conserved sequence of FXX (F/Y)(S/T)(Y/F). In mammals, this hydrophobic motif is FPQFSY. The serine or threonine residue in this motif must also be phosphorylated to activate Kinase activity of Akt. This is also a conserved residue (Ref.1).
Activation of Akt can begin with several events, mainly the binding of a Ligand to a Receptor in the cell membrane. Most common Ligands activating Akt include Growth factors, Cytokines, Mitogens and Hormones. Insulin and a variety of Growth factors bind to RTK (Receptor Tyrosine Kinase) and cause autophosphorylation of tyrosine residues on the intracellular domain of the receptor. PI3K (Phosphoinositol 3-Kinase) is recruited to the phosphotyrosine residues (consensus sequence pYXXM) via SH2 domains in the regulatory domain (p85), and is therefore targeted to the inner cell membrane. Binding of the p85 subunit of PI3K to the phosphorylated RTK leads to conformational changes in the catalytic domain of PI3K (p110) and consequent kinase activation. PI3K can be activated by Ras. Insulin can also activate PI3K via IRS1 (Insulin Receptor Substrate-1). GPCR (G-Protein-Coupled Receptor) also activates PI3K through GN-Beta (Guanine Nucleotide-Binding Protein-Beta) and GN-Gamma (Guanine Nucleotide-Binding Protein-Gamma) subunits of G-proteins. Cytokines can also activate PI3K via JAK1 (Janus Kinase-1). In B-Cells, PI3K is activated by BCR (B-Cell Receptor) via SYK (Spleen Tyrosine Kinase) and BCAP (B-Cell Receptor Associated Protein). PI3K then phosphorylates membrane bound PIP2 to generate PIP3. The binding of PIP3 to the PH domain anchors Akt to the plasma membrane and allows its phosphorylation and activation by PDK1 (Phosphoinositide-Dependent Kinase-1). DNA-PK, CDC37 (Cell Division Cycle-37), HSP90 (Heat Shock Protein-90KD) and PKCƒ{Beta (Protein Kinase-C-Beta) are also reported to phosphorylate Akt. Integrins also activates Akt via FAK (Focal Adhesion Kinase), Paxillin and ILK (Integrin-Linked Kinase). Akt can also be activated in response to a variety of cellular stress, such as heat shock, administration of ultra violet light, ischemia (a decrease in blood supply), hypoxia (oxygen deficiency), hypoglycemia (abnormally low level of glucose in the blood) and oxidative stress. The activity of Akt is negatively regulated by PTEN (Phosphatase and Tensin Homolog), SHIP (SH2-Containing Inositol Phosphatase) and CTMP (Carboxyl-Terminal Modulator Protein) (Ref.2, 3 & 4).
The actions of Akt in the cell are numerous and diverse, but all result in anti-apoptosis, or pro-cell proliferation effects. These physiological roles of Akt include involvement in metabolism, protein synthesis, apoptosis pathways, transcription factor regulation and the cell cycle. Akt exerts its effects in the cell by phosphorylating a variety of downstream substrates. The downstream targets of Akt include BAD (BCL2 Antagonist of Cell Death), Caspase9, FKHR (Forkhead Transcriptional Factor), GLUTs (Glucose Transporters), eNOS (Nitric Oxide Synthase), PFK2 (6-Phosphofructo-2-Kinase), PFK1(6-Phosphofructo-Kinase), mTOR (Mammalian Target of Rapamycin), IKK (I-KappaB Kinase), NF-KappaB (Nuclear Factor-KappaB), GSK3 (Glycogen Synthase Kinase-3), WNK1(WNK Lysine deficient Protein Kinase-1), PRAS40 (Proline-Rich Akt Substrate 40 kDa), p47Phox, YAP (Yes-Associated Protein-1), Htt (Huntingtin), Ataxin, AR (Androgen Receptor), ASK1 (Apoptosis Signal-Regulating Kinase-1), MDM2 (Mouse Double Minute-2), CREB (cAMP Response Element-Binding Protein), p21CIP1 (Cyclin Dependent Kinase Inhibitor-p21), p27KIP1 (Cyclin Dependent Kinase Inhibitor-p27) , Chk1 (Cell Cycle Checkpoint Kinase-1), XIAP (X-Linked Inhibitor of Apoptosis Protein), Raf1 (v-Raf1 Murine Leukemia Viral Oncogene Homolog-1), PDE3B (Phosphodiesterase 3B cGMP-Inhibited), TSC (Tuberous Sclerosis Gene) and GABA(A)R (Gamma-Aminobutyric Acid Receptor-A) (Ref.4).
Akt inhibits apoptosis by phosphorylating the BAD component of the BAD/BclXL (Bcl2 Related Protein Long Isoform) complex. Phosphorylated BAD binds to 14-3-3 causing dissociation of the BAD/BclXL complex and allowing cell survival. Akt activates IKK, which ultimately leads to NF-KappaB activation and cell survival. Other direct targets of Akt are members of the FKHRL1 (Forkhead-Related Family of Mammalian Transcription Factor-1). In the presence of survival factors, Akt1 phosphorylates FKHRL1, leading to the association of FKHRL1 with 14-3-3 proteins and its retention in the cytoplasm. Survival factor withdrawal leads to FKHRL1 dephosphorylation, nuclear translocation, and target gene activation. Within the nucleus, FKHRL1 most likely triggers apoptosis by inducing the expression of genes that are critical for cell death, such as the TNFSF6 (Tumor Necrosis Factor Ligand Superfamily Member-6) gene. Another notable substrate of Akt is the death protease Caspase9. Phosphorylation of Caspase9 decreases apoptosis by directly inhibiting the protease activity. Akt also activates TERT (Telomere Reverse Transcriptase), which is responsible for telomere maintenance and DNA stability. Akt has been linked to angiogenesis, through the activation of eNOS, which influences long-term blood vessel growth. Akt can regulate several levels of Glucose metabolism. It enhances Glucose-uptake in Insulin-responsive tissues by inducing the expression of GLUT1 and GLUT3 and the translocation of GLUT4 to the plasma membrane; the GLUTs transport glucose into the cell. Akt also activates Glycogen synthesis by phosphorylating and inactivating GSK3, which leads to the activation of Glycogen Synthase and CyclinD1. Akt phosphorylates PDE3B on Ser273. This activates PDE3B and results in regulation of intracellular levels of cyclic nucleotides in response to Insulin. Akt induces glycolysis through the phosphorylation and activation PFK2, which in turn activates PFK1. These enzymes convert Fructose-6-Phosphate into Fructose-1, 6-Bisphosphate, a key step in Glucose metabolism. Akt may also be involved in activation of the nutrient-dependent Thr/Ser kinase, mTOR. Activation of mTOR results in the phosphorylation of ribosomal protein S6 kinase, p70S6K. Akt also phosphorylates the two tumor suppressor genes TSC1 and TSC2, which are negative regulators of the mTOR-S6K pathway. Phosphorylation of TSC1 and TSC2 results in suppression of their inhibitory activity and may also target the proteins for degradation. Activation of mTOR also results in phosphorylation and inactivation of eIF4EBP (Eukaryotic Initiation Factor-4E Binding Protein), an inhibitor of the translation initiation factor eIF4E. Nonphosphorylated PHASI binds to eIF4E (Eukaryotic Initiation Factor-4E) and inhibits protein synthesis. Akt also phosphorylates GAB2 (GRB2-Associated Binding Protein-2) on Ser159. Phosphorylation of Ser159 on Gab2 by Akt/PKB appears to negatively regulate GAB2 tyrosine phosphorylation by the ErbB receptor tyrosine kinases, although the underlying mechanism has not been solved (Ref.5 & 6).
The transcription factor CREB is directly phosphorylated at Ser133 by Akt. This causes an increased affinity of CREB for its co-activator protein, CRB (Crumbs). The heterodimer, now an active transcription factor, promotes transcription of genes that contain CREs (cAMP responsive elements) in their promoter, such as the anti-apoptotic genes Bcl2 and Mcl1. Akt also phosphorylates AR at two serine residues, Ser210 and Ser270, which causes a decrease in AR activity on the p21 promoter. In addition to causing cell cycle progression, this also results in apoptosis inhibition in certain cell types, through other actions of AR. YAP is another transcription factor that is phosphorylated by Akt, and is of importance because it does not contain an Akt consensus sequence. Akt phosphorylates Ser127 on YAP, which causes association with 14-3-3 proteins, nuclear export and cytoplasmic localization. Akt has also been shown to phosphorylate p21 directly, on Thr145. p21 is a member of the Cip/Kip family of CDK inhibitors that arrest the cell cycle and therefore limit cell proliferation. p21 can also promote cell cycle progression, via mediating the assembly and activity of cyclin D1-CDK4/6 complexes. P27 is another cyclin-dependent kinase inhibitor, of the Kip family. P27 inhibits CDK2 and CDK4/6 complexes, which is located in the nuclear localization signal. NLS targets protein to nucleus via nuclear import machinery, and phosphorylation in this region of p27 results in nuclear exclusion. 14-3-3 proteins bind phosphorylated p27 and cause active export from nucleus. Without p27 in the nucleus, the cyclin-CDK complexes form and promote cell cycle progression. Akt also phosphorylates MDM2. MDM2 is phosphorylated at many sites, only two of which have been identified. Ser166 is phosphorylated by Akt. Akt phosphorylation of MDM2 allows its entry into the nucleus where it targets p53 for degradation (Ref.7, 8, 9 & 10).
PRAS40 is a 40 kDa substrate of AKT. Activated AKT phosphorylates PRAS40 on threonine 246, enabling PRAS40 to bind to 14-3-3. AKT and PRAS40 are components of the PI3K pathway. This pathway plays a role in glucose uptake, cell growth, and apoptosis inhibition. The precise function of PRAS40 is not yet known; however, it has been hypothesized that PRAS40 interacts with SH3 and WW domain containing proteins, and may change the function of these proteins. Akt phosphorylates, both in vitro and in vivo, the GABA(A)R, the principal receptor mediating fast inhibitory synaptic transmission in the mammalian brain. Akt-mediated phosphorylation increases the number of GABA(A)Rs on the plasma membrane surface, thereby increasing the receptor-mediated synaptic transmission in neurons. XIAP is a physiological substrate of Akt. Akt interacts with and phosphorylates XIAP at serine 87. Phosphorylation of XIAP by Akt inhibits both its autoubiquitination and cisplatin-induced ubiquitination. These effects reduce XIAP degradation and the increased levels of XIAP are associated with decreased cisplatin-stimulated Caspase3 activity and programmed cell death. Htt is also a substrate of Akt and phosphorylation of Htt by Akt is crucial to mediate the neuroprotective effects of IGF1 (Insulin-Like Growth Factor-I). WNK1 is a physiologically relevant target of Insulin signaling through PI3K and Akt and functions as a negative regulator of Insulin-stimulated mitogenesis (Ref.11, 12 & 13). Akt also phosphorylates Ataxin1 and modulate neurodegeration.14-3-3 protein mediates the neurotoxicity of Ataxin1 by binding to and stabilizing Ataxin1, thereby slowing its normal degradation. Akt also decreases ASK1 kinase activity by phosphorylating a consensus Akt site at serine 83 of ASK1. Akt also interacts with the JIP1 (JNK Interacting Protein-1) scaffold and inhibits the ability of JIP1 to form active JNK signaling complexes. The binding of Akt to JIP1 is isoform specific; Akt1 but not Akt2 interacts with JIP1. Thus, Akt can inhibit one or more steps within the JNK signaling pathway, depending on the complement of components that form the functional JNK signaling module. Akt mediates PI3K-dependent p47Phox phosphorylation, which contributes to respiratory burst activity in human neutrophils. AKT impair Chk1 through phosphorylation, ubiquitination, and reduced nuclear localization to promote genomic instability in tumor cells. Akt and its upstream regulators are deregulated in a wide range of solid tumors and hematologic malignancies, hence the Akt pathway is considered a key determinant of biologic aggressiveness of these tumors, and a major potential target for novel anti-cancer therapies (Ref.14 & 15).
References
- 1
- Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, Wang Y, Liu F, Wang YT. Control of synaptic strength, a novel function of Akt.
- 2
- Pommery N, Henichart JP. Involvement of PI3K/Akt pathway in prostate cancer--potential strategies for developing targeted therapies.
- 3
- Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function.
- 4
- Fornaro M, Plescia J, Chheang S, Tallini G, Zhu YM, King M, Altieri DC, Languino LR. Fibronectin protects prostate cancer cells from tumor necrosis factor-alpha-induced apoptosis via the AKT/survivin pathway.
- 5
- Georgakis GV, Younes A. From Rapa Nui to rapamycin: targeting PI3K/Akt/mTOR for cancer therapy.
- 6
- Jin ZH, Kurosu T, Yamaguchi M, Arai A, Miura O. Hematopoietic cytokines enhance Chk1-dependent G2/M checkpoint activation by etoposide through the Akt/GSK3 pathway to inhibit apoptosis.
- 7
- Fu M, Rao M, Wu K, Wang C, Zhang X, Hessien M, Yeung YG, Gioeli D, Weber MJ, Pestell RG. The androgen receptor acetylation site regulates cAMP and AKT but not ERK-induced activity.
- 8
- Min YH, Cheong JW, Kim JY, Eom JI, Lee ST, Hahn JS, Ko YW, Lee MH. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia.
- 9
- Li Y, Dowbenko D, Lasky LA. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival.
- 10
- Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis.
 关于我们
关于我们